| Title: | Dynamic Multi-Species Size Spectrum Modelling |
| Date: | 2026-07-17 |
| Type: | Package |
| Description: | A set of classes and methods to set up and run multi-species, trait based and community size spectrum ecological models, focused on the marine environment. |
| Maintainer: | Gustav Delius <gustav.delius@york.ac.uk> |
| Version: | 3.2.0 |
| License: | GPL-3 |
| Imports: | assertthat, deSolve, dplyr, ggplot2 (≥ 3.4.0), ggrepel, grid, lubridate, methods, plotly, plyr, progress, Rcpp, reshape2, rlang, lifecycle, pak |
| LinkingTo: | Rcpp |
| Depends: | R (≥ 3.5) |
| Suggests: | testthat (≥ 3.0.0), withr, vdiffr, diffviewer, roxygen2, knitr, rmarkdown, quarto, pkgdown, covr, spelling |
| Collate: | 'age_mat.R' 'helpers.R' 'MizerParams-class.R' 'MizerSim-class.R' 'registerExtensions.R' 'ArraySpeciesBySize-class.R' 'ArrayTimeBySpecies-class.R' 'ArrayTimeBySpeciesBySize-class.R' 'ArrayResourceBySize-class.R' 'generic_methods.R' 'background.R' 'reproduction.R' 'saveParams.R' 'species_params.R' 'getRequiredRDD.R' 'setColours.R' 'setInteraction.R' 'setPredKernel.R' 'setSearchVolume.R' 'setMaxIntakeRate.R' 'setMetabolicRate.R' 'setMetadata.R' 'setExtMort.R' 'setExtEncounter.R' 'diffusion.R' 'second_order_w.R' 'setExtDiffusion.R' 'setReproduction.R' 'setResource.R' 'setFishing.R' 'setInitialValues.R' 'setBevertonHolt.R' 'upgrade.R' 'selectivity_funcs.R' 'pred_kernel_funcs.R' 'resource_dynamics.R' 'resource_semichemostat.R' 'resource_logistic.R' 'numerical_methods.R' 'transport.R' 'project_n.R' 'project.R' 'mizer-package.R' 'project_methods.R' 'rate_functions.R' 'summary_methods.R' 'indicator_functions.R' 'plots.R' 'plotBiomassObservedVsModel.R' 'plotYieldObservedVsModel.R' 'animateSpectra.R' 'newMultispeciesParams.R' 'wrapper_functions.R' 'newSingleSpeciesParams.R' 'steady.R' 'extension.R' 'data.R' 'RcppExports.R' 'deprecated.R' 'get_initial_n.R' 'compareParams.R' 'customFunction.R' 'manipulate_species.R' 'calibrate.R' 'scaleRates.R' 'match.R' 'matchGrowth.R' 'steadySingleSpecies.R' 'defaults_edition.R' 'validSpeciesParams.R' 'zzz.R' |
| Encoding: | UTF-8 |
| LazyData: | true |
| URL: | https://sizespectrum.org/mizer/, https://github.com/sizespectrum/mizer |
| BugReports: | https://github.com/sizespectrum/mizer/issues |
| Language: | en-GB |
| RdMacros: | lifecycle |
| VignetteBuilder: | knitr, quarto |
| Config/testthat/edition: | 3 |
| Config/roxygen2/version: | 8.0.0 |
| NeedsCompilation: | yes |
| Packaged: | 2026-07-19 22:30:02 UTC; gustav |
| Author: | Gustav Delius  [cre, aut, cph],
Finlay Scott [aut, cph],
Julia Blanchard
[aut, cph],
Ken Andersen
[aut, cph],
Richard Southwell [ctb, cph]
[cre, aut, cph],
Finlay Scott [aut, cph],
Julia Blanchard
[aut, cph],
Ken Andersen
[aut, cph],
Richard Southwell [ctb, cph] |
| Repository: | CRAN |
| Date/Publication: | 2026-07-19 22:50:02 UTC |
mizer: Multi-species size-based modelling in R
Description
The mizer package implements multi-species size-based modelling in R. It has been designed for modelling marine ecosystems.
Details
Using mizer is relatively simple. There are three main stages:
-
Setting the model parameters. This is done by creating an object of class MizerParams. This includes model parameters such as the life history parameters of each species, and the range of the size spectrum. There are several setup functions that help to create a MizerParams objects for particular types of models:
-
Running a simulation. This is done by calling the
project()function with the model parameters. This produces an object of MizerSim that contains the results of the simulation. -
Exploring results. After a simulation has been run, the results can be explored using a range of plotting_functions, summary_functions and indicator_functions.
See the mizer website for full details of the principles behind mizer and how the package can be used to perform size-based modelling.
Author(s)
Maintainer: Gustav Delius gustav.delius@york.ac.uk (ORCID) [copyright holder]
Authors:
Gustav Delius gustav.delius@york.ac.uk (ORCID) [copyright holder]
Finlay Scott drfinlayscott@gmail.com [copyright holder]
Julia Blanchard julia.blanchard@utas.edu.au (ORCID) [copyright holder]
Ken Andersen kha@aqua.dtu.dk (ORCID) [copyright holder]
Other contributors:
Richard Southwell richard.southwell@york.ac.uk [contributor, copyright holder]
See Also
Useful links:
Report bugs at https://github.com/sizespectrum/mizer/issues
Check that a rate function returns the correct output dimensions
Description
Called by setRateFunction() to verify that a candidate rate function
returns an array (or vector/list) of the correct dimensions for the
requested rate.
Usage
.checkRateFunctionOutput(params, rate, fun)
Arguments
params |
A MizerParams object |
rate |
Name of the rate being replaced, e.g. |
fun |
Name of the candidate function to validate. |
Value
Invisibly NULL. Called for its side-effect of stopping with an
informative error if the output has the wrong shape.
S3 class for resource size spectra
Description
Several functions in mizer return a vector over the full size grid holding
a resource-related quantity such as the resource number density, the
resource mortality, the intrinsic resource birth rate or carrying capacity.
The ArrayResourceBySize class wraps these vectors to provide convenient
print(), summary(), plot(), and as.data.frame() methods.
Usage
ArrayResourceBySize(x, value_name = NULL, units = NULL, params = NULL)
is.ArrayResourceBySize(x)
Arguments
x |
A numeric vector over the full size grid. For
|
value_name |
A string giving the human-readable name for the value. |
units |
A string giving the units (e.g. "1/year"). |
params |
A |
Details
An ArrayResourceBySize object behaves just like a regular numeric vector
for arithmetic operations and subsetting. It carries three lightweight
attributes:
-
value_name– a human-readable name for the value (e.g. "Resource mortality"). -
units– the units of the value (e.g. "1/year"). -
params– theMizerParamsobject that the value was computed from.
Value
An ArrayResourceBySize object (inherits from numeric).
is.ArrayResourceBySize() returns TRUE if x is an
ArrayResourceBySize object, FALSE otherwise.
See Also
print(), summary(), as.data.frame(), plot()
Examples
mort <- getResourceMort(NS_params)
is.ArrayResourceBySize(mort)
summary(mort)
plot(mort)
S3 class for species x size rate arrays
Description
Many functions in mizer return two-dimensional arrays (species x size)
holding rates like encounter rate, feeding level, growth rate, mortality etc.
The ArraySpeciesBySize class wraps these arrays to provide convenient
print(), summary(), plot(), and as.data.frame() methods.
Usage
ArraySpeciesBySize(
x,
value_name = NULL,
units = NULL,
params = NULL,
representation = c("point", "average")
)
is.ArraySpeciesBySize(x)
Arguments
x |
A matrix (species x size). For |
value_name |
A string giving the human-readable name for the value. |
units |
A string giving the units (e.g. "g/year", "1/year"). |
params |
A |
representation |
Either |
Details
An ArraySpeciesBySize object behaves just like a regular matrix for
arithmetic operations and subsetting. It carries two lightweight attributes:
-
value_name– a human-readable name for the value (e.g. "Encounter rate"). -
units– the units of the rate (e.g. "g/year").
Value
An ArraySpeciesBySize object (inherits from matrix and array).
is.ArraySpeciesBySize() returns TRUE if x is an
ArraySpeciesBySize object, FALSE otherwise.
See Also
print(), summary(), as.data.frame(), plot()
Examples
enc <- getEncounter(NS_params)
is.ArraySpeciesBySize(enc)
summary(enc)
S3 class for time x resource-size arrays
Description
The NResource() function returns a two-dimensional array (time x size)
holding the resource number density through time. The
ArrayTimeByResourceBySize class wraps this array to provide convenient
print(), summary(), plot(), and as.data.frame() methods.
Usage
ArrayTimeByResourceBySize(x, value_name = NULL, units = NULL, params = NULL)
is.ArrayTimeByResourceBySize(x)
Arguments
x |
A matrix (time x size). For |
value_name |
A string giving the human-readable name for the value. |
units |
A string giving the units (e.g. "1/g"). |
params |
A |
Details
An ArrayTimeByResourceBySize object behaves just like a regular matrix for
arithmetic operations and subsetting. It carries these lightweight
attributes:
-
value_name– a human-readable name for the value (e.g. "Number density"). -
units– the units of the value (e.g. "1/g"). -
params– theMizerParamsobject that the value was computed from.
Value
An ArrayTimeByResourceBySize object (inherits from matrix and
array).
is.ArrayTimeByResourceBySize() returns TRUE if x is an
ArrayTimeByResourceBySize object, FALSE otherwise.
See Also
print(), summary(), as.data.frame(), plot()
Examples
nr <- NResource(NS_sim)
is.ArrayTimeByResourceBySize(nr)
summary(nr)
plot(nr)
S3 class for time x species arrays
Description
Some functions in mizer return two-dimensional arrays (time x species)
holding quantities like biomass, abundance, or yield rate through time.
The ArrayTimeBySpecies class wraps these arrays to provide convenient
print(), summary(), plot(), and as.data.frame() methods.
Usage
ArrayTimeBySpecies(x, value_name = NULL, units = NULL, params = NULL)
is.ArrayTimeBySpecies(x)
Arguments
x |
A matrix (time x species). For |
value_name |
A string giving the human-readable name for the value. |
units |
A string giving the units (e.g. "g", "g/year"). |
params |
A |
Details
An ArrayTimeBySpecies object behaves just like a regular matrix for
arithmetic operations and subsetting. It carries these lightweight attributes:
-
value_name– a human-readable name for the value (e.g. "Biomass"). -
units– the units of the value (e.g. "g", "g/year"). -
params– theMizerParamsobject that created the values.
Value
An ArrayTimeBySpecies object (inherits from matrix and array).
is.ArrayTimeBySpecies() returns TRUE if x is an
ArrayTimeBySpecies object, FALSE otherwise.
See Also
print(), summary(), as.data.frame(), plot()
Examples
bio <- getBiomass(NS_sim)
is.ArrayTimeBySpecies(bio)
summary(bio)
S3 class for time x species x size arrays
Description
Some functions in mizer return three-dimensional arrays (time x species x
size) holding quantities like fishing mortality, feeding level, or predation
mortality through time. The ArrayTimeBySpeciesBySize class wraps these
arrays to provide convenient print(), summary(), plot(),
animate(), and as.data.frame() methods.
Usage
ArrayTimeBySpeciesBySize(
x,
value_name = NULL,
units = NULL,
params = NULL,
representation = c("point", "average")
)
is.ArrayTimeBySpeciesBySize(x)
Arguments
x |
A 3D array (time x species x size). For
|
value_name |
A string giving the human-readable name for the value. |
units |
A string giving the units (e.g. "1/year"). |
params |
A |
representation |
Either |
Details
An ArrayTimeBySpeciesBySize object behaves just like a regular array for
arithmetic operations and subsetting. It carries these lightweight attributes:
-
value_name– a human-readable name for the value (e.g. "Fishing mortality"). -
units– the units of the value (e.g. "1/year"). -
params– theMizerParamsobject that the value was computed from.
Value
An ArrayTimeBySpeciesBySize object (inherits from array).
is.ArrayTimeBySpeciesBySize() returns TRUE if x is an
ArrayTimeBySpeciesBySize object, FALSE otherwise.
See Also
print(), summary(), as.data.frame(), plot(),
animateSpectra()
Examples
fmort <- getFMort(NS_sim)
is.ArrayTimeBySpeciesBySize(fmort)
summary(fmort)
plot(fmort, time = 2007)
Beverton Holt function to calculate density-dependent reproduction rate
Description
Takes the density-independent rates R_{di} of egg production (as
calculated by getRDI()) and returns
reduced, density-dependent reproduction rates R_{dd} given as
R_{dd} = R_{di}
\frac{R_{max}}{R_{di} + R_{max}}
where
R_{max} are the maximum possible reproduction rates that must be
specified in a column in the species parameter dataframe.
(All quantities in the above equation are species-specific but we dropped
the species index for simplicity.)
Usage
BevertonHoltRDD(rdi, species_params, ...)
Arguments
rdi |
Vector of density-independent reproduction rates
|
species_params |
A species parameter dataframe. Must contain a column
|
... |
Unused |
Details
This is only one example of a density-dependence. You can write your own
function based on this example, returning different density-dependent
reproduction rates. Three other examples provided are RickerRDD(),
SheperdRDD(), noRDD() and constantRDD(). For more explanation see
setReproduction().
Value
Vector of density-dependent reproduction rates.
See Also
Other functions calculating density-dependent reproduction rate:
RickerRDD(),
SheperdRDD(),
constantEggRDI(),
constantRDD(),
noRDD()
Alias for set_multispecies_model()
Description
![[Deprecated]](./figures/lifecycle-deprecated.svg) An alias provided for backward compatibility with mizer version <= 1.0
An alias provided for backward compatibility with mizer version <= 1.0
Usage
MizerParams(
species_params,
interaction = matrix(1, nrow = nrow(species_params), ncol = nrow(species_params)),
min_w_pp = 1e-10,
min_w = 0.001,
max_w = NULL,
no_w = 100,
n = 2/3,
q = 0.8,
f0 = 0.6,
kappa = 1e+11,
lambda = 2 + q - n,
r_pp = 10,
...
)
Arguments
species_params |
A data frame of species-specific parameter values. |
interaction |
Optional interaction matrix of the species (predator species x prey species). By default all entries are 1. See "Setting interaction matrix" section below. |
min_w_pp |
The smallest size of the resource spectrum. By default this is set to the smallest value at which any of the consumers can feed. |
min_w |
Sets the size of the eggs of all species for which this is not
given in the |
max_w |
The largest size of the consumer spectrum. By default this is
set to the largest |
no_w |
The number of size bins in the consumer spectrum. |
n |
The allometric growth exponent. This can be overruled for individual
species by including a |
q |
Allometric exponent of search volume |
f0 |
Expected average feeding level. Used to set |
kappa |
The coefficient |
lambda |
Used to set power-law exponent for resource capacity if the
|
r_pp |
|
... |
Further arguments passed to |
Details
If species_params contains a w_inf column then it is copied to w_max.
If max_w is not supplied then it is set to 1.1 * max(species_params$w_max).
The supplied min_w_pp is shifted up by one grid step before being passed
to newMultispeciesParams() to compensate for the fact that newer mizer
versions extend the full size grid below min_w_pp.
Missing legacy columns in species_params are filled as follows:
gear = species, k = 0, alpha = 0.6, erepro = 1,
sel_func = "knife_edge", knife_edge_size = w_mat if needed,
catchability = 1, ks = h * 0.2, and m = 1.
If h is missing it is calculated from k_vb, alpha, f0 and w_max.
If gamma is missing it is calculated from f0, h, beta, sigma,
lambda and kappa.
Value
A MizerParams object
A class to hold the parameters for a size based model.
Description
Although it is possible to build a MizerParams object by hand it is
not recommended and several constructors are available. Dynamic simulations
are performed using project() function on objects of this class. As a
user you should never need to access the slots inside a MizerParams object
directly.
Details
The MizerParams class is fairly complex with a large number of
slots, many of which are multidimensional arrays. The dimensions of these
arrays is strictly enforced so that MizerParams objects are consistent
in terms of number of species and number of size classes.
The MizerParams class does not hold any dynamic information, e.g.
abundances or harvest effort through time. These are held in
MizerSim objects.
Slots
metadataA list with metadata information. See
setMetadata().mizer_versionThe package version of mizer (as returned by
packageVersion("mizer")) that created or upgraded the model.extensionsDescribes the extension chain needed to run the model. The entries are named by extension identifier (also the S4 marker class name) and ordered in S3 dispatch order, from outermost to innermost extension. It is either a named character vector whose values are requirement strings (version strings, installation specifications, or
NA_character_), or a named list whose entries are length-2 character vectorsc(requirement = ..., version = ...). Theversionrecords the version of the extension package that last upgraded the object (NAif unknown) and is used byneeds_upgrading(). UserecordExtension()to write entries rather than modifying the slot directly. Extension subclasses are marker classes only and must not add slots.time_createdA POSIXct date-time object with the creation time.
time_modifiedA POSIXct date-time object with the last modified time.
wThe size grid for the fish part of the spectrum. An increasing vector of weights (in grams) running from the smallest egg size to the largest maximum size.
dwThe widths (in grams) of the size bins
w_fullThe size grid for the full size range including the resource spectrum. An increasing vector of weights (in grams) running from the smallest resource size to the largest maximum size of fish. The last entries of the vector have to be equal to the content of the w slot.
dw_fullThe width of the size bins for the full spectrum. The last entries have to be equal to the content of the dw slot.
w_min_idxA vector holding the index of the weight of the egg size of each species
maturityAn array (species x size) that holds the proportion of individuals of each species at size that are mature. This enters in the calculation of the spawning stock biomass with
getSSB(). Set withsetReproduction().psiAn array (species x size) that holds the allocation to reproduction for each species at size,
\psi_i(w). Changed withsetReproduction().intake_maxAn array (species x size) that holds the maximum intake for each species at size. Changed with
setMaxIntakeRate().search_volAn array (species x size) that holds the search volume for each species at size. Changed with
setSearchVolume().metabAn array (species x size) that holds the metabolism for each species at size. Changed with
setMetabolicRate().mu_bAn array (species x size) that holds the external mortality rate
\mu_{ext.i}(w). Changed withsetExtMort().ext_encounterAn array (species x size) that holds the external encounter rate
E_{ext.i}(w). Changed withsetExtEncounter().ext_diffusionAn array (species x size) that holds the external rate at which the abundance density is redistributed over body size due to mixing, beyond the deterministic growth dynamics. Changed with
ext_diffusion().pred_kernelAn array (species x predator size x prey size) that holds the predation coefficient of each predator at size on each prey size. If this is NA then the following two slots will be used. Changed with
setPredKernel().ft_pred_kernel_eAn array (species x log of predator/prey size ratio) that holds the Fourier transform of the feeding kernel in a form appropriate for evaluating the encounter rate integral. If this is NA then the
pred_kernelwill be used to calculate the available energy integral. Changed withsetPredKernel().ft_pred_kernel_pAn array (species x log of predator/prey size ratio) that holds the Fourier transform of the feeding kernel in a form appropriate for evaluating the predation mortality integral. If this is NA then the
pred_kernelwill be used to calculate the integral. Changed withsetPredKernel().ft_pred_kernel_dAn array (species x log of predator/prey size ratio) that holds the Fourier transform of the feeding kernel in a form appropriate for evaluating the predation-diffusion integral (used when
use_predation_diffusionisTRUE). It differs fromft_pred_kernel_eonly whensecond_order_w[["bin_average"]]isTRUE, where it carries the extra power of prey size that the diffusion integrand needs; otherwise it equalsft_pred_kernel_e. Changed withsetPredKernel().rr_ppA vector the same length as the w_full slot. The size specific growth rate of the resource spectrum.
cc_ppA vector the same length as the w_full slot. The size specific carrying capacity of the resource spectrum.
resource_dynamicsName of the function for projecting the resource abundance density by one timestep.
other_dynamicsA named list of functions for projecting the values of other dynamical components of the ecosystem that may be modelled by a mizer extensions you have installed. The names of the list entries are the names of those components.
other_encounterA named list of functions for calculating the contribution to the encounter rate from each other dynamical component.
other_mortA named list of functions for calculating the contribution to the mortality rate from each other dynamical components.
other_paramsA list containing the parameters needed by any mizer extensions you may have installed to model other dynamical components of the ecosystem.
rates_funcsA named list with the names of the functions that should be used to calculate the rates needed by
project(). By default this will be set to the names of the built-in rate functions.sc![[Experimental]](./figures/lifecycle-experimental.svg) The community abundance of the scaling community
The community abundance of the scaling communityspecies_paramsA data.frame to hold the species specific parameters. See
species_params()for details.given_species_paramsA data.frame to hold the species parameters that were given explicitly rather than obtained by default calculations.
gear_paramsData frame with parameters for gear selectivity. See
setFishing()for details.interactionThe species specific interaction matrix,
\theta_{ij}. Changed withsetInteraction().selectivityAn array (gear x species x w) that holds the selectivity of each gear for species and size,
S_{g,i,w}. Changed withsetFishing().catchabilityAn array (gear x species) that holds the catchability of each species by each gear,
Q_{g,i}. Changed withsetFishing().initial_effortA vector containing the initial fishing effort for each gear. Changed with
setFishing().initial_nAn array (species x size) that holds the initial abundance of each species at each weight.
initial_n_ppA vector the same length as the w_full slot that describes the initial resource abundance at each weight.
initial_n_otherA list with the initial abundances of all other ecosystem components. Has length zero if there are no other components.
resource_paramsList with parameters for resource.
A-
Formerly used to flag background species via
NAvalues. Replaced by theis_backgroundcolumn inspecies_params. Will be removed in a future version. linecolourA named vector of colour values, named by species. Used to give consistent colours in plots.
linetypeA named vector of linetypes, named by species. Used to give consistent line types in plots.
ft_maskAn array (species x w_full) with zeros for weights larger than the maximum weight of each species. Used to efficiently minimize wrap-around errors in Fourier transform calculations.
use_predation_diffusionA logical flag controlling whether predation-induced diffusion is included when calculating rates with
mizerDiffusion(). Defaults toFALSEto preserve the behaviour of previous mizer versions. Set toTRUEto enable the diffusion term from the jump-growth equation.second_order_wA named list with entry
flux(the advective flux scheme:"upwind","van_leer", or"centred") and logical entrybin_average(controls whether bin-averaging is used for rates). Both default to the first-order setting to preserve the behaviour of previous mizer versions.
See Also
project() MizerSim()
emptyParams() newMultispeciesParams()
newCommunityParams()
newTraitParams()
Constructor for the MizerSim class
Description
A constructor for the MizerSim class. This is used by
project() to create MizerSim objects of the right
dimensions. It is not necessary for users to use this constructor.
Usage
MizerSim(params, t_dimnames = NA, t_max = 100, t_save = 1)
Arguments
params |
a MizerParams object |
t_dimnames |
Numeric vector that is used for the time dimensions of the slots. Default = NA. |
t_max |
The maximum time step of the simulation. Only used if t_dimnames = NA. Default value = 100. |
t_save |
How often should the results of the simulation be stored. Only used if t_dimnames = NA. Default value = 1. |
Value
An object of type MizerSim
A class to hold the results of a simulation
Description
A class that holds the results of projecting a MizerParams
object through time using project().
Details
A new MizerSim object can be created with the MizerSim()
constructor, but you will never have to do that because the object is
created automatically by project() when needed.
As a user you should never have to access the slots of a MizerSim object
directly. Instead there are a range of functions to extract the information.
N() and NResource() return arrays with the saved abundances of
the species and the resource population at size respectively. getEffort()
returns the fishing effort of each gear through time.
getTimes() returns the vector of times at which simulation results
were stored and idxFinalT() returns the index with which to access
specifically the value at the final time in the arrays returned by the other
functions. getParams() extracts the ecosystem state as a MizerParams
object with initial abundances set to values from the simulation; finalParams()
and initialParams() are convenient shorthands for the final and initial
time steps. There are also several
summary_functions and plotting_functions
available to explore the contents of a MizerSim object.
The arrays all have named dimensions. The names of the time dimension
denote the time in years. The names of the w dimension are weights in grams
rounded to three significant figures. The names of the sp dimension are the
same as the species name in the order specified in the species_params data
frame. The names of the gear dimension are the names of the gears, in the
same order as specified when setting up the MizerParams object.
Extensions of mizer can use the n_other slot to store the abundances of
other ecosystem components and these extensions should provide their own
functions for accessing that information.
The MizerSim class has changed since previous versions of mizer. To use
a MizerSim object created by a previous version, you need to upgrade it
with validSim().
Slots
paramsAn object of type MizerParams. If this params object uses extensions, the
MizerSimobject uses the same extension chain viaparams@extensions;MizerSimhas no separate extension slot.nThree-dimensional array (time x species x size) that stores the projected community number densities.
n_ppAn array (time x size) that stores the projected resource number densities.
n_otherA list array (time x component) that stores the projected values for other ecosystem components.
effortAn array (time x gear) that stores the fishing effort by time and gear.
sim_paramsA named list of the parameters passed to
project()orprojectToSteady()to produce this simulation, such asmethodanddt.
Time series of size spectra
Description
Fetch the simulation results for the size spectra over time.
Usage
N(sim)
NResource(sim)
Arguments
sim |
A MizerSim object |
Value
For N(): An ArrayTimeBySpeciesBySize object (time x species x
size) with the number density of consumers.
For NResource(): An array (time x size) with the number density of resource
Examples
str(N(NS_sim))
str(NResource(NS_sim))
Time series of other components
Description
Fetch the simulation results for other components over time.
Usage
NOther(sim)
finalNOther(sim)
Arguments
sim |
A MizerSim object |
Value
For NOther: A list array indexed by time and component that stores the projected
values for other ecosystem components.
For finalNOther: A named list holding the values of other ecosystem components at the
end of the simulation
See Also
Other extension tools:
clearExtensionChain(),
coerceToExtensionClass(),
getRegisteredExtensions(),
initialNOther<-(),
recordExtension(),
registerExtension(),
registerExtensions(),
setComponent(),
setRateFunction()
Example interaction matrix for the North Sea example
Description
The interaction coefficient between predator and prey species in the North Sea.
Usage
NS_interaction
Format
A 12 x 12 matrix.
Source
Blanchard et al.
Examples
params <- newMultispeciesParams(NS_species_params_gears,
interaction = NS_interaction)
Example MizerParams object for the North Sea example
Description
A MizerParams object created from the NS_species_params_gears species
parameters and the inter interaction matrix together with an initial
condition corresponding to the steady state obtained from fishing with an
effort
effort = c(Industrial = 0, Pelagic = 1, Beam = 0.5, Otter = 0.5).
Usage
NS_params
Format
A MizerParams object
Source
Blanchard et al.
See Also
Other example parameter objects:
NS_sim
Examples
sim = project(NS_params, effort = c(Industrial = 0, Pelagic = 1,
Beam = 0.5, Otter = 0.5))
plot(sim)
Example MizerSim object for the North Sea example
Description
A MizerSim object containing a simulation with historical fishing mortalities from the North Sea, as created in the tutorial "A Multi-Species Model of the North Sea".
Usage
NS_sim
Format
A MizerSim object
Source
https://sizespectrum.org/mizer/articles/a_multispecies_model_of_the_north_sea.html
See Also
Other example parameter objects:
NS_params
Examples
plotBiomass(NS_sim)
Example species parameter set based on the North Sea
Description
This data set is based on species in the North Sea (Blanchard et al.). It is
a data.frame that contains all the necessary information to be used by the
MizerParams() constructor. As there is no gear column, each species is
assumed to be fished by a separate gear.
Usage
NS_species_params
Format
A data frame with 12 rows and 7 columns. Each row is a species.
- species
Name of the species
- w_max
Computational upper size boundary, defaulting to
1.5 * w_inf.- w_mat
Size at maturity
- beta
Size preference ratio
- sigma
Width of the size-preference
- R_max
Maximum reproduction rate
- k_vb
The von Bertalanffy k parameter
- w_inf
The von Bertalanffy asymptotic size. This is the required maximum-size parameter.
Source
Blanchard et al.
Examples
params <- newMultispeciesParams(NS_species_params)
Example species parameter set based on the North Sea with different gears
Description
This data set is based on species in the North Sea (Blanchard et al.).
It is similar to the data set NS_species_params except that
this one has an additional column specifying the fishing gear that
operates on each species.
Usage
NS_species_params_gears
Format
A data frame with 12 rows and 8 columns. Each row is a species.
- species
Name of the species
- w_max
Computational upper size boundary, defaulting to
1.5 * w_inf.- w_mat
Size at maturity
- beta
Size preference ratio
- sigma
Width of the size-preference
- R_max
Maximum reproduction rate
- k_vb
The von Bertalanffy k parameter
- w_inf
The von Bertalanffy asymptotic size. This is the required maximum-size parameter.
- gear
Name of the fishing gear
Source
Blanchard et al.
Examples
params <- MizerParams(NS_species_params_gears)
Ricker function to calculate density-dependent reproduction rate
Description
Takes the density-independent rates R_{di} of egg production and
returns reduced, density-dependent rates R_{dd} given as
R_{dd} = R_{di} \exp(- b R_{di})
Usage
RickerRDD(rdi, species_params, ...)
Arguments
rdi |
Vector of density-independent reproduction rates
|
species_params |
A species parameter dataframe. Must contain a column
|
... |
Unused |
Value
Vector of density-dependent reproduction rates.
See Also
Other functions calculating density-dependent reproduction rate:
BevertonHoltRDD(),
SheperdRDD(),
constantEggRDI(),
constantRDD(),
noRDD()
Sheperd function to calculate density-dependent reproduction rate
Description
Takes the density-independent rates R_{di} of egg production and returns
reduced, density-dependent rates R_{dd} given as
R_{dd} = \frac{R_{di}}{1+(b\ R_{di})^c}
Usage
SheperdRDD(rdi, species_params, ...)
Arguments
rdi |
Vector of density-independent reproduction rates
|
species_params |
A species parameter dataframe. Must contain columns
|
... |
Unused |
Details
With b = 1/R_{max} and c = 1 this reduces to the Beverton-Holt
reproduction rate, see BevertonHoltRDD().
Value
Vector of density-dependent reproduction rates.
See Also
Other functions calculating density-dependent reproduction rate:
BevertonHoltRDD(),
RickerRDD(),
constantEggRDI(),
constantRDD(),
noRDD()
Add lines to an existing plot
Description
addPlot() adds another set of values to an existing ggplot. The first
method supports adding an ArraySpeciesBySize object to a compatible plot,
for example to compare the same rate before and after a model change.
The method checks whether the existing plot uses a compatible x variable,
and warns if the y variable or y-axis units appear to differ.
Usage
addPlot(
plot,
x,
species = NULL,
total = FALSE,
background = TRUE,
colour = NULL,
linetype = "dashed",
linewidth = 0.8,
alpha = 1,
...
)
Arguments
plot |
A ggplot2 object to which the new values should be added. |
x |
An object containing the values to add. |
species |
Character vector of species to include. |
total |
A boolean value that determines whether the total over all
selected species is plotted as well. Default is |
background |
A boolean value that determines whether background species
are included. Ignored if the model does not contain background species.
Default is |
colour |
Optional fixed colour for the added lines. If |
linetype |
Optional fixed line type for the added lines. If |
linewidth |
Width of the added lines. |
alpha |
Transparency of the added lines. |
... |
Further arguments used by only some of the methods: For
For
|
Value
A ggplot2 object.
See Also
Other plotting functions:
animate(),
plot,
plot2(),
plotBiomass(),
plotCDF(),
plotCDF2(),
plotDiet(),
plotFMort(),
plotFeedingLevel(),
plotGrowthCurves(),
plotMizerParams,
plotMizerSim,
plotPredMort(),
plotRelative(),
plotSpectra(),
plotSpectra2(),
plotSpectraRelative(),
plotYield(),
plotYieldGear(),
plotting_functions
Examples
p <- plot(getEncounter(NS_params), species = "Cod")
addPlot(p, getEncounter(NS_params), species = "Cod")
Add new species
Description
Takes a MizerParams object and adds additional species with given parameters to the ecosystem. It sets the initial values for these new species to their steady-state solution in the given initial state of the existing ecosystem. This will be close to the true steady state if the abundances of the new species are sufficiently low. Hence the abundances of the new species are set so that they are at most 1/100th of the resource power law. Their reproductive efficiencies are set so as to keep them at that low level.
Usage
addSpecies(
params,
species_params,
gear_params = data.frame(),
initial_effort,
interaction,
steady = TRUE,
info_level = 3,
...
)
Arguments
params |
A mizer params object for the original system. |
species_params |
Data frame with the species parameters of the new species we want to add to the system. |
gear_params |
Data frame with the gear parameters for the new species. If not provided then the new species will not be fished. |
initial_effort |
A named vector with the effort for any new fishing gear
introduced in |
interaction |
Interaction matrix. A square matrix giving either the interaction coefficients between all species or only those between the new species. In the latter case all interaction between an old and a new species are set to 1. If this argument is missing, all interactions involving a new species are set to 1. |
steady |
If |
info_level |
Controls the amount of information messages that are shown when the function sets default values for parameters. Higher levels lead to more messages. Set to 0 to suppress all such messages. |
... |
Currently unused. |
Details
The resulting MizerParams object will use the same size grid where possible, but if one of the new species needs a larger range of w (either because a new species has an egg size smaller than those of existing species or a maximum size larger than those of existing species) then the grid will be expanded and all arrays will be enlarged accordingly.
If any of the rate arrays of the existing species had been set by the user to values other than those calculated as default from the species parameters, then these will be preserved. Only the rates for the new species will be calculated from their species parameters.
After adding the new species, the background species are not retuned and
the system is not run to steady state. This could be done with steady().
The new species will have a reproduction level of 1/4, this can then be
changed with setBevertonHolt()
Value
An object of type MizerParams
See Also
removeSpecies(), renameSpecies()
Examples
params <- newTraitParams()
species_params <- data.frame(
species = "Mullet",
w_max = 173,
w_mat = 15,
beta = 283,
sigma = 1.8,
h = 30,
a = 0.0085,
b = 3.11
)
params <- addSpecies(params, species_params)
plotSpectra(params)
Adjust the size grid
Description
This function adjusts the size grid in a MizerParams object to the desired minimum and maximum size. It can both expand and truncate the grid. If the grid is truncated, any data outside the new grid is discarded. A warning is issued if there is non-negligible biomass in the discarded size bins.
Usage
adjustSizeGrid(params, ...)
## S3 method for class 'MizerParams'
adjustSizeGrid(
params,
new_min_w = min(params@species_params$w_min),
new_max_w = max(params@species_params$w_max),
new_min_w_pp = min(params@w_full),
preserve_species = params@species_params$species,
tol = 1e-06,
...
)
Arguments
params |
A MizerParams object. |
... |
Additional arguments. |
new_min_w |
The new minimum size in the grid. Defaults to the minimum egg size of all species. |
new_max_w |
The new maximum size in the grid. Defaults to the maximum asymptotic size of all species. |
new_min_w_pp |
The new minimum size of the resource spectrum. Defaults
to the current minimum of |
preserve_species |
A vector of species names for which all rate arrays should be copied over to the new params object rather than being re-calculated from the species parameters. If missing, all species are preserved. |
tol |
A numeric value specifying the tolerance for truncation losses.
The following checks are made separately for each species and a warning is
raised, listing the affected species, if the lost fraction exceeds this
value for any of them: the fraction of the species' biomass lost, the
fraction of the diet of the smallest individuals of the species lost to
resource truncation, and the fraction of the diet of the largest
individuals of the species lost to resource truncation. Defaults to
|
Value
A new MizerParams object with the updated size grid.
Calculate age at maturity
Description
Uses the size-dependent growth rate and the size at maturity to calculate the age at maturity.
Usage
age_mat(params, ...)
Arguments
params |
A MizerParams object |
... |
Currently unused. |
Details
Using that by definition of the growth rate g(w) = dw/dt we have that
\mathrm{age_{mat}} = \int_0^{w_{mat}.}\frac{dw}{g(w)}
In the implementation this integral is approximated on the model size grid by
summing dw / g(w) over all size bins with w < w_mat.
Value
A named vector. The names are the species names and the values are the ages at maturity.
Examples
age_mat(NS_params)
Calculate age at maturity from von Bertalanffy growth parameters
Description
This is not a good way to determine the age at maturity because the von Bertalanffy growth curve is not reliable for larvae and juveniles. However this was used in previous versions of mizer and is supplied for backwards compatibility.
Usage
age_mat_vB(object, ...)
Arguments
object |
A MizerParams object or a species_params data frame |
... |
Currently unused. |
Details
Uses the age at maturity that is implied by the von Bertalanffy growth curve
specified by the w_inf, k_vb, t0, a and b parameters in the
species_params data frame.
If any of k_vb is missing for a species, the function returns NA for that
species. Default values of b = 3 and t0 = 0 are used if these are
missing. If w_inf is missing, w_max is used instead.
Value
A named vector. The names are the species names and the values are the ages at maturity.
Animate size-dependent quantities through time
Description
Creates an interactive plotly animation in which a play button steps through time, drawing one line per species at each frame.
Usage
animate(
x,
species = NULL,
log_x = TRUE,
log_y = TRUE,
log = NULL,
wlim = c(NA, NA),
llim = c(NA, NA),
ylim = c(NA, NA),
tlim = c(NA, NA),
size_axis = c("w", "l"),
total = FALSE,
background = TRUE,
frame_duration = 500,
transition_duration = frame_duration,
easing = "linear",
...
)
animateSpectra(sim, ...)
Arguments
x |
A |
species |
Name or vector of names of the species to be plotted. By default all species are plotted. |
log_x |
If |
log_y |
If |
log |
A character string specifying which axes to log-transform:
|
wlim |
A numeric vector of length two providing lower and upper limits
for the body-size (x) axis. Use |
llim |
A numeric vector of length two providing lower and upper limits
for the length (x) axis when |
ylim |
A numeric vector of length two providing lower and upper limits
for the value (y) axis. Use |
tlim |
A numeric vector of length two providing lower and upper limits
for the animated time window, e.g. |
size_axis |
Whether to plot size as weight ( |
total |
A boolean value that determines whether the total over all
selected species is plotted as an additional trace called |
background |
A boolean value that determines whether background species
are included. Ignored if the model does not contain background species.
Default is |
frame_duration |
Duration in milliseconds for which each saved frame is displayed. Default is 500. |
transition_duration |
Duration in milliseconds of the interpolation
between frames. Use |
easing |
The Plotly easing function to use when interpolating between
frames. Default is |
... |
Further arguments used by only some of the methods: For
|
sim |
A |
Details
The function dispatches on the class of x:
-
MizerSim— animates the community abundance spectra (number density or biomass density vs body size). Resource, background species, and a community total can be added via theresource,background, andtotalarguments. Thepowerargument controls whether the y-axis shows number density (power = 0), biomass density (power = 1, default), or biomass density in logarithmic size bins (power = 2). Both axes are log10 by default and can each be switched to linear withlog_x = FALSEorlog_y = FALSE. -
ArrayTimeBySpeciesBySize— animates any per-species, size-resolved quantity returned by aMizerSimaccessor, such asgetFMort(),getFeedingLevel(), orgetPredMort(). Both axes are log10 by default and can each be switched to linear withlog_x = FALSEorlog_y = FALSE. Background species and a species total can be added via thebackgroundandtotalarguments.
Species linecolours and linetypes follow params@linecolour and
params@linetype.
animateSpectra() is retained as a backward-compatible alias.
Value
A plotly object with one animated line trace per plotted group. Use the play button or the slider to step through time.
See Also
Other plotting functions:
addPlot(),
plot,
plot2(),
plotBiomass(),
plotCDF(),
plotCDF2(),
plotDiet(),
plotFMort(),
plotFeedingLevel(),
plotGrowthCurves(),
plotMizerParams,
plotMizerSim,
plotPredMort(),
plotRelative(),
plotSpectra(),
plotSpectra2(),
plotSpectraRelative(),
plotYield(),
plotYieldGear(),
plotting_functions
Examples
# Animate biomass density spectra, showing only sizes above 0.1 g
animate(NS_sim, power = 2, wlim = c(0.1, NA), tlim = c(1997, 2007))
# Animate fishing mortality through time
animate(getFMort(NS_sim))
# Animate feeding level for two species only
animate(getFeedingLevel(NS_sim), species = c("Cod", "Herring"))
Restrict plot data to a range of weights
Description
Internal helper that filters a plot data frame to the weight range given by
wlim. It is exported so that extension packages (such as mizerMR) can reuse
it in their own array plot() methods.
Usage
apply_wlim(data, wlim)
Arguments
data |
A data frame with a numeric |
wlim |
A length-2 numeric vector giving the lower and upper weight
limits. Either entry may be |
Value
The subset of data with w inside wlim.
Convert mizer arrays to data frames
Description
The as.data.frame() methods for mizer array classes turn matrix- and
array-like results into tidy long-form data frames, with one row per
observed combination of species, size and/or time. The numeric result is
always stored in a column called value.
Usage
## S3 method for class 'ArraySpeciesBySize'
as.data.frame(x, row.names = NULL, optional = FALSE, ...)
## S3 method for class 'ArrayTimeBySpecies'
as.data.frame(x, row.names = NULL, optional = FALSE, ...)
## S3 method for class 'ArrayTimeBySpeciesBySize'
as.data.frame(x, row.names = NULL, optional = FALSE, ...)
Arguments
x |
An |
row.names |
Optional and included only for compatibility with the base
generic. |
optional |
Optional and included only for compatibility with the base
generic. A logical value. If |
... |
Further arguments. They are currently ignored by the mizer methods. |
Details
The returned columns are:
-
ArraySpeciesBySize:w,value,Species. -
ArrayTimeBySpecies:time,value,Species. -
ArrayTimeBySpeciesBySize:time,Species,w,value.
If the original object has non-numeric or missing dimension names, sequential
indices are used for the time or w columns. Species names are taken from
the row, column or dimension names of the original object.
Value
A data frame in long format.
See Also
print(), summary(), str(), plot(), ArraySpeciesBySize(),
ArrayTimeBySpecies(), ArrayTimeBySpeciesBySize()
Examples
enc <- getEncounter(NS_params)
head(as.data.frame(enc))
biomass <- getBiomass(NS_sim)
head(as.data.frame(biomass))
Assert that an object's extension chain is compatible with the session
Description
Stops with an informative error if the object's extension chain is not a
suffix of the session's registered maximal chain, or (when check_class is
TRUE) if the object does not inherit from the expected S4 marker class.
Usage
assertExtensionChain(
object,
extensions = objectExtensions(object),
check_class = TRUE
)
Arguments
object |
A |
extensions |
Named character vector giving the object's extension chain.
Defaults to |
check_class |
Logical. If |
Value
Invisibly TRUE. Called for its side-effect of stopping on
incompatibility.
Strip extension classes from a mizer object
Description
Coerces a MizerParams or MizerSim object back to its plain base class,
removing any S4 extension marker classes. For MizerSim, also strips the
extension class from the embedded params slot.
Usage
baseMizerClass(object)
Arguments
object |
A |
Value
The same object coerced to MizerParams or MizerSim.
Bin-average a summary-integral weight when second-order is enabled
Description
Convenience wrapper around bin_average_weight() that is gated on the
bin_average entry of the model's second_order_w slot. When second-order
bin-averaging is switched off (the default), the weight K is returned
unchanged so that the summary functions reproduce the previous left-edge
Riemann sums byte-for-byte. When it is switched on, the trapezoidal
bin-average of the weight is returned.
Usage
bin_average_summary_weight(K, params)
Arguments
K |
A numeric vector of weights indexed over the size grid, or a numeric matrix with the size dimension running along the columns. |
params |
A MizerParams object whose |
Value
The weight K, bin-averaged when params@second_order_w[["bin_average"]]
is TRUE, otherwise returned unchanged.
Trapezoidal bin-average of a per-bin weight
Description
Internal helper for the second-order summary integrals. A summary
diagnostic \int N(w) K(w)\, dw is discretised on the finite-volume
grid as \sum_j N_j \bar K_j \Delta w_j, where N_j is the cell
average of the density over bin [w_j, w_{j+1}]. To be second order in
the bin width the point weight K(w_j) must be replaced by the bin
average
\bar K_j = \frac{1}{\Delta w_j}\int_{w_j}^{w_{j+1}} K(w)\,dw
\approx \tfrac12\big(K(w_j) + K(w_{j+1})\big).
The trapezoidal average \tfrac12(K_j + K_{j+1}) is uniformly second
order and exact whenever K is linear in w (e.g. the first
moment K = w, for which it equals (w_{j+1}^2 - w_j^2)/(2\Delta
w_j)).
Usage
bin_average_weight(K)
Arguments
K |
A numeric vector of weights indexed over the size grid, or a numeric array whose last dimension runs over the size grid (e.g. a species-by-size matrix or a gear-by-species-by-size array). |
Details
The weight K is supplied already evaluated on the size grid (a vector
indexed over the bins, or a matrix with the size dimension running along the
columns). The top bin has no right-hand neighbour on the grid, so its weight
is left unaveraged (one-sided); the density there is negligible, so this
does not affect the second-order accuracy of the totals.
This helper is shared with the reproduction integrals (issue #376), which also need the trapezoidal bin-average of a composite weight.
Value
An object of the same shape as K containing the trapezoidal
bin-averaged weights.
Geometric bin centres of the size grid
Description
Internal helper for the second-order plotting code. A finite-volume cell
average N_j = (1/\Delta w_j)\int_{w_j}^{w_{j+1}} N\,dw does not live at
the left bin edge w_j but at the geometric bin centre
w^*_j = \sqrt{w_j\,w_{j+1}} = w_j\sqrt{\beta},
where \beta = w_{j+1}/w_j is the (constant) bin ratio of the
logarithmic grid. This is the log-symmetric, second-order-correct location at
which to plot a bin-averaged quantity (it is exact for the community spectrum
N\propto w^{-2}). It is a uniform half-bin shift to the right on the log
axis, the same for the consumer grid w and the full prey/resource grid
w_full.
Usage
bin_midpoints(params, w_full = FALSE)
Arguments
params |
A MizerParams object. |
w_full |
If |
Value
A numeric vector of geometric bin centres, one per grid node.
Box predation kernel
Description
A predation kernel where the predator/prey mass ratio is uniformly distributed on an interval.
Usage
box_pred_kernel(ppmr, ppmr_min, ppmr_max)
Arguments
ppmr |
A vector of predator/prey size ratios |
ppmr_min |
Minimum predator/prey mass ratio |
ppmr_max |
Maximum predator/prey mass ratio |
Details
Writing the predator mass as w and the prey mass as w_p, the
feeding kernel is 1 if w/w_p is between ppmr_min and
ppmr_max inclusive and zero otherwise. ppmr_min must be strictly smaller
than ppmr_max. The parameters need to be given in the species parameter
dataframe in the columns ppmr_min and ppmr_max.
Value
A vector giving the value of the predation kernel at each of the
predator/prey mass ratios in the ppmr argument.
See Also
Other predation kernel:
lognormal_pred_kernel(),
power_law_pred_kernel(),
truncated_lognormal_pred_kernel()
Examples
params <- NS_params
# Set all required paramters before changing kernel type
species_params(params)$ppmr_max <- 4000
species_params(params)$ppmr_min <- 200
species_params(params)$pred_kernel_type <- "box"
plot(w_full(params), getPredKernel(params)["Cod", 10, ], type="l", log="x")
Calculate selectivity from gear parameters
Description
This function calculates the selectivity for each gear, species and size from
the gear parameters. It is called by setFishing() when the selectivity is
not set by the user. The returned array is initialised to zero, so
gear-species combinations that are not listed in gear_params(params) remain
zero. For each listed combination the function named in sel_func is called
with w = params@w, the corresponding species parameters, and the
selectivity parameters from the matching row in gear_params(params).
Usage
calc_selectivity(params)
Arguments
params |
A MizerParams object |
Value
An array (gear x species x size) with the selectivity values
Bin-averaged selectivity
By default the selectivity is point-sampled at the grid nodes params@w,
i.e. at the left edge of each size bin. This is only first-order accurate in
the bin size when the selectivity is used in the finite-volume update of the
size spectrum. When the bin_average entry of the second_order_w() slot is
TRUE, each selectivity function is instead integrated over its size bin,
so that selectivity[g, i, j] holds the bin average
\bar S_{g,i,j} = \frac{1}{\Delta w_j} \int_{w_j}^{w_{j+1}} S_{g,i}(w)\, dw.
The integral is evaluated with a composite-midpoint rule on a log-spaced sub-grid of each bin, mirroring the bin-integrated predation kernel. This lifts the fishing mortality towards second order at no extra runtime cost (the integration happens once here, the rate functions are unchanged). A welcome side effect is that a knife-edge gear then gets the exact fraction of the straddling bin that lies above the knife edge, removing a grid artefact.
Examples
params <- NS_params
str(calc_selectivity(params))
calc_selectivity(params)["Pelagic", "Herring", ]
Calibrate the model scale to match total observed biomass
Description
Given a MizerParams object params for which biomass observations are
available for at least some species via the biomass_observed column in the
species_params data frame, this function returns an updated MizerParams
object which is rescaled with scaleModel() so that the total biomass in
the model agrees with the total observed biomass.
Usage
calibrateBiomass(params, ...)
Arguments
params |
A MizerParams object |
... |
Additional arguments passed to the method. |
Details
Biomass observations usually only include individuals above a certain size. This size should be specified in a biomass_cutoff column of the species parameter data frame. If this is missing, it is assumed that all sizes are included in the observed biomass, i.e., it includes larval biomass.
After using this function the total biomass in the model will match the
total biomass, summed over all species. However the biomasses of the
individual species will not match observations yet, with some species
having biomasses that are too high and others too low. So after this
function you may want to use matchBiomasses(). This is described in the
blog post at https://blog.mizer.sizespectrum.org/posts/2021-08-20-a-5-step-recipe-for-tuning-the-model-steady-state/.
If you have observations of the yearly yield instead of biomasses, you can
use calibrateYield() instead of this function.
Value
A MizerParams object. If no non-missing observed biomass values are provided, the original object is returned unchanged.
Examples
params <- NS_params
species_params(params)$biomass_observed <-
c(0.8, 61, 12, 35, 1.6, 20, 10, 7.6, 135, 60, 30, 78)
species_params(params)$biomass_cutoff <- 10
params2 <- calibrateBiomass(params)
plotBiomassObservedVsModel(params2)
Calibrate the model scale to match total observed number
Description
Given a MizerParams object params for which number observations are
available for at least some species via the number_observed column in the
species_params data frame, this function returns an updated MizerParams
object which is rescaled with scaleModel() so that the total number in
the model agrees with the total observed number.
Usage
calibrateNumber(params, ...)
Arguments
params |
A MizerParams object |
... |
Additional arguments passed to the method. |
Details
Number observations usually only include individuals above a certain size. This size should be specified in a number_cutoff column of the species parameter data frame. If this is missing, it is assumed that all sizes are included in the observed number, i.e., it includes larval number.
After using this function the total number in the model will match the
total number, summed over all species. However the numbers of the
individual species will not match observations yet, with some species
having numbers that are too high and others too low. So after this
function you may want to use matchNumbers(). This is described in the
blog post at https://blog.mizer.sizespectrum.org/posts/2021-08-20-a-5-step-recipe-for-tuning-the-model-steady-state/.
If you have observations of the yearly yield instead of numbers, you can
use calibrateYield() instead of this function.
Value
A MizerParams object. If no non-missing observed number values are provided, the original object is returned unchanged.
Examples
params <- NS_params
species_params(params)$number_observed <-
c(0.8, 61, 12, 35, 1.6, 20, 10, 7.6, 135, 60, 30, 78)
species_params(params)$number_cutoff <- 10
params2 <- calibrateNumber(params)
Calibrate the model scale to match total observed yield
Description
Usage
calibrateYield(params, ...)
Arguments
params |
A MizerParams object |
... |
Additional arguments passed to the method. |
Details
This function has been deprecated and will be removed in the future unless you have a use case for it. If you do have a use case for it, please let the developers know by creating an issue at https://github.com/sizespectrum/mizer/issues.
Given a MizerParams object params for which yield observations are
available for at least some species via the yield_observed column in the
species_params data frame, this function returns an updated MizerParams
object which is rescaled with scaleModel() so that the total yield in
the model agrees with the total observed yield.
After using this function the total yield in the model will match the
total observed yield, summed over all species. However the yields of the
individual species will not match observations yet, with some species
having yields that are too high and others too low. So after this
function you may want to use matchYields().
If you have observations of species biomasses instead of yields, you can
use calibrateBiomass() instead of this function.
Value
A MizerParams object. If no non-missing observed yield values are provided, the original object is returned unchanged.
Examples
params <- NS_params
species_params(params)$yield_observed <-
c(0.8, 61, 12, 35, 1.6, 20, 10, 7.6, 135, 60, 30, 78)
gear_params(params)$catchability <-
c(1.3, 0.065, 0.31, 0.18, 0.98, 0.24, 0.37, 0.46, 0.18, 0.30, 0.27, 0.39)
params2 <- calibrateYield(params)
plotYieldObservedVsModel(params2)
Y-axis label for a cumulative-distribution plot
Description
Y-axis label for a cumulative-distribution plot
Usage
cdf_y_label(power, normalise)
Arguments
power |
The power of weight that the abundance was multiplied by. |
normalise |
Whether the cumulative distribution is normalised. |
Value
A character string for the y-axis label.
Clear the registered extension chain
Description
Clears the session's extension registry. You can then create a new
extension chain with registerExtensions().
Usage
clearExtensionChain()
Value
Invisibly, an empty character vector.
See Also
"Using mizer extension packages":
vignette("using-extension-packages", package = "mizer")
Other extension tools:
NOther(),
coerceToExtensionClass(),
getRegisteredExtensions(),
initialNOther<-(),
recordExtension(),
registerExtension(),
registerExtensions(),
setComponent(),
setRateFunction()
Coerce a mizer object to its registered extension class
Description
Coerces a MizerParams or MizerSim object to the S4 marker class
corresponding to the object's own extension chain. For MizerSim, the
extension chain is read from sim@params@extensions.
Usage
coerceToExtensionClass(object, extensions = objectExtensions(object))
Arguments
object |
A |
extensions |
Optional extension chain. Defaults to the chain stored in
|
Value
The same object coerced to the appropriate marker class, or to the base class for an empty extension chain.
See Also
"Creating a mizer extension package":
vignette("creating-extension-packages", package = "mizer")
Other extension tools:
NOther(),
clearExtensionChain(),
getRegisteredExtensions(),
initialNOther<-(),
recordExtension(),
registerExtension(),
registerExtensions(),
setComponent(),
setRateFunction()
Compare two extension chains
Description
Compare two extension chains
Usage
compareExtensionChains(old, new)
Arguments
old |
Named character vector for the previously registered chain. |
new |
Named character vector for the proposed chain. |
Value
One of "identical", "new_is_suffix", "old_is_suffix", or
"incompatible".
Compare two MizerParams objects and print out differences
Description
Compare two MizerParams objects and print out differences
Usage
compareParams(params1, params2, ...)
Arguments
params1 |
First MizerParams object |
params2 |
Second MizerParams object |
... |
Additional arguments passed to the method. |
Value
Invisibly returns a character vector of difference messages, one element per difference. As a side effect, prints the differences in a human-readable format.
Examples
params1 <- NS_params
params2 <- params1
species_params(params2)$w_mat[1] <- 10
compareParams(params1, params2)
Alias for validSpeciesParams()
Description
An alias provided for backward compatibility with mizer version <= 2.5.2
Usage
completeSpeciesParams(species_params)
Arguments
species_params |
The user-supplied species parameter data frame |
Details
validGivenSpeciesParams() checks the validity of the given species
parameters. It throws an error if
the
speciescolumn does not exist or contains duplicatesthe asymptotic size
w_infis not specified for all species (but see the backwards-compatibility note below)
If a weight-based parameter is missing but the corresponding length-based
parameter is given, as well as the a and b parameters for length-weight
conversion, then the weight-based parameters are added. If both length and
weight are given, then weight is used and an info_about_default condition
is signalled if the two are inconsistent.
The required maximum-size parameter is w_inf, the von Bertalanffy
asymptotic size of an average individual. For backwards compatibility, if no
w_inf column is given, its values are taken from the w_repro_max column
if that is present, or otherwise from the w_max column, and an
informational message is issued. (w_repro_max is preferred over w_max
because in earlier versions of mizer it was the size at which growth stopped
and is therefore the closest analogue to the asymptotic size.)
Some inconsistencies in the size parameters are resolved as follows:
Any
w_matthat is not smaller thanw_infis set tow_inf / 4.Any
w_mat25that is not smaller thanw_matis set to NA.Any
w_minthat is not smaller thanw_matis set to0.001orw_mat /10, whichever is smaller.Any
w_repro_maxthat is not larger thanw_matis set to4 * w_mat.
The row names of the returned data frame will be the species names.
If species_params was provided as a tibble it is converted back to an
ordinary data frame.
The function tests for some typical misspellings of parameter names, like wrong capitalisation or missing underscores and issues a warning if it detects such a name.
validSpeciesParams() first calls validGivenSpeciesParams() but then
goes further by adding default values for species parameters that were not
provided. It only sets defaults for those species parameters that are not
owned by a single rate-setting function, namely those that are read by
several of them (n), that are used only when projecting (alpha), that
determine the size grid (w_min, w_max), that are needed for the
length-weight conversion (a, b) or that are used only for reporting
(is_background). The function sets default values if any of the following
species parameters are missing or NA:
-
w_maxis set to1.5 * w_inf(it is only a computational boundary) -
w_repro_maxis set tow_inf -
w_matis set tow_inf/4 -
w_minis set to0.001 -
alphais set to0.6 -
nis set to3/4 -
ais set to0.01 -
bis set to3 -
is_backgroundis set toFALSE
All other species parameters are given their default values by the
rate-setting function that uses them, so that each default has a single
home. For example p and k are set by setMetabolicRate(), z_ext, d
and z0 by setExtMort(), E_ext by setExtEncounter(), D_ext by
setExtDiffusion(), interaction_resource by setInteraction(), beta
and sigma by setPredKernel(), q and gamma by setSearchVolume(),
and erepro, m, w_mat25 and R_max by setReproduction(). These
columns are therefore absent from the data frame returned by
validSpeciesParams() but present in the species parameters of a
MizerParams object, because setParams() calls all the rate-setting
functions.
Note that the species parameters returned by these functions are not
guaranteed to produce a viable model. More checks of the parameters are
performed by the individual rate-setting functions (see setParams() for the
list of these functions).
Value
For validSpeciesParams(): A valid species parameter data frame with
additional parameters with default values.
For validGivenSpeciesParams(): A valid species parameter data frame
without additional parameters.
See Also
species_params(), validGearParams(), validParams(), validSim()
Choose egg production to keep egg density constant
Description
The new egg production is set to compensate for the loss of individuals from
the smallest size class through growth and mortality. The result should not
be modified by density dependence, so this should be used together with
the noRDD() function, see example.
Usage
constantEggRDI(params, n, e_growth, mort, diffusion, ...)
Arguments
params |
A MizerParams object |
n |
A matrix of species abundances (species x size). |
e_growth |
A two dimensional array (species x size) holding the energy
available for growth as calculated by |
mort |
A two dimensional array (species x size) holding the mortality
rate as calculated by |
diffusion |
A two dimensional array (species x size) holding the
diffusion rate as calculated by |
... |
Unused |
Value
Vector with the value for each species
See Also
Other functions calculating density-dependent reproduction rate:
BevertonHoltRDD(),
RickerRDD(),
SheperdRDD(),
constantRDD(),
noRDD()
Examples
# choose an example params object
params <- NS_params
# We set the reproduction rate functions
params <- setRateFunction(params, "RDI", "constantEggRDI")
params <- setRateFunction(params, "RDD", "noRDD")
# Now the egg density should stay fixed no matter how we fish
sim <- project(params, effort = 10, progress_bar = FALSE)
# To check that indeed the egg densities have not changed, we first construct
# the indices for addressing the egg densities
no_sp <- nrow(params@species_params)
idx <- (params@w_min_idx - 1) * no_sp + (1:no_sp)
# Now we can check equality between egg densities at the start and the end
all.equal(finalN(sim)[idx], initialN(params)[idx])
Give constant reproduction rate
Description
Simply returns the value from species_params$constant_reproduction.
Usage
constantRDD(rdi, species_params, ...)
Arguments
rdi |
Vector of density-independent reproduction rates
|
species_params |
A species parameter dataframe. Must contain a column
|
... |
Unused |
Value
Vector species_params$constant_reproduction
See Also
Other functions calculating density-dependent reproduction rate:
BevertonHoltRDD(),
RickerRDD(),
SheperdRDD(),
constantEggRDI(),
noRDD()
Helper function to keep other components constant
Description
Helper function to keep other components constant
Usage
constant_other(params, n_other, component, ...)
Arguments
params |
MizerParams object |
n_other |
Abundances of other components |
component |
Name of the component that is being updated |
... |
Unused |
Value
The current value of the component
Convert plotting data from weight to length
Description
When size_axis = "l", adds a length column l computed from the weight
column w using each species' weight-length relationship. For
size_axis = "w" the data is returned unchanged.
Usage
convert_plot_size_axis(
plot_dat,
params,
size_axis,
species_col = "Species",
drop_w = TRUE
)
Arguments
plot_dat |
A data frame of plotting data with a |
params |
A MizerParams object providing the weight-length parameters. |
size_axis |
Either |
species_col |
Name of the column identifying the species. Default is
|
drop_w |
If |
Value
The plotting data, with a length column l added (and w optionally
dropped) when size_axis = "l".
Replace a mizer function with a custom version
Description
This function allows you to make arbitrary changes to how mizer works by
allowing you to replace any mizer function with your own version. You
should do this only as a last resort, when you find that you can not use
the standard mizer extension mechanism to achieve your goal.
Usage
customFunction(name, fun)
Arguments
name |
Name of mizer function to replace |
fun |
The custom function to use as replacement |
Details
If the function you need to overwrite is one of the mizer rate functions,
then you should use setRateFunction() instead of this function. Similarly
you should use resource_dynamics()<- to change the resource dynamics and
setReproduction() to change the density-dependence in reproduction.
You should also investigate whether you can achieve your goal by introducing
additional ecosystem components with setComponent().
If you find that your goal really does require you to overwrite a mizer function, please also create an issue on the mizer issue tracker at https://github.com/sizespectrum/mizer/issues to describe your goal, because it will be interesting to the mizer community and may motivate future improvements to the mizer functionality.
Note that customFunction() only overwrites the function used by the mizer
code. It does not overwrite the function that is exported by mizer. This
will become clear when you run the code in the Examples section.
This function does not in any way check that your replacement function is compatible with mizer. Calling this function can totally break mizer. However you can always undo the effect by reloading mizer with
detach(package:mizer, unload = TRUE) library(mizer)
Value
No return value, called for side effects
See Also
"Extending mizer":
vignette("extending-mizer", package = "mizer")
Examples
## Not run:
fake_project <- function(...) "Fake"
customFunction("project", fake_project)
mizer::project(NS_params) # This will print "Fake"
project(NS_params) # This will still use the old project() function
# To undo the effect:
customFunction("project", project)
mizer::project(NS_params) # This will again use the old project()
## End(Not run)
Set defaults for predation kernel parameters
Description
If the predation kernel type has not been specified for a species, then it
is set to "lognormal" and the default values are set for the parameters
beta and sigma.
Usage
default_pred_kernel_params(object)
Arguments
object |
Either a MizerParams object or a species parameter data frame |
Value
The object with updated columns in the species params data frame.
Default editions
Description
Function to set and get which edition of default choices is being used.
Usage
defaults_edition(edition = NULL)
Arguments
edition |
NULL or a numerical value. |
Details
The mizer functions for creating new models make a lot of choices for default values for parameters that are not provided by the user. Sometimes we find better ways to choose the defaults and update mizer accordingly. When we do this, we will increase the edition number.
If you call defaults_edition() without an argument it returns the
currently active edition. Otherwise it sets the active edition to the
given value.
Users who want their existing code for creating models not to change behaviour when run with future versions of mizer should explicitly set the desired defaults edition at the top of their code.
The most recent edition is edition 2. It will become the default in the next release. The current default is edition 1. The following defaults are changed in edition 2:
-
catchability= 0.3 instead of 1 -
initial effort= 1 instead of 0 -
gammais set to ensure a feeding level off0for larvae with the current value ofinteraction_resourceinstead of interaction_resource = 1'. -
initial_nis set usingget_steady_state_n()instead of the rather arbitrary old choice. In
setReproduction(),psiis no longer forced to 1 abovew_repro_max; its value is determined entirely by the maturity ogive and the reproductive proportion.
Value
If edition is NULL, the currently active edition number. If
edition is supplied, the function sets the global
mizer_defaults_edition option, emits a message, and returns the supplied
value invisibly.
Define S4 marker classes for a set of dispatch extensions
Description
Creates a linear inheritance chain of S4 classes: the outermost extension
extends the next, which extends the next, down to the base MizerParams /
MizerSim class. Existing classes are checked for compatibility instead of
being redefined.
Usage
defineExtensionClasses(extensions)
Arguments
extensions |
Named character vector of extensions (full chain or dispatch subset). Non-dispatch entries are silently ignored. |
Value
Invisibly, the named character vector of dispatch extensions.
Define an S4 class or verify it extends the expected parent
Description
If class does not yet exist, defines it as a virtual-free S4 class that
contains parent, registered in .GlobalEnv. If class already exists,
stops with an error unless it already extends parent.
Usage
defineOrCheckClass(class, parent)
Arguments
class |
Character string — the S4 class name to define or check. |
parent |
Character string — the required parent class. |
Value
Invisibly, class.
Check whether two objects are different
Description
Check whether two objects are numerically different, ignoring all attributes.
Usage
different(a, b)
Arguments
a |
First object |
b |
Second object |
Details
We use this helper function in particular to see if a new value for a slot in MizerParams is different from the existing value in order to give the appropriate messages.
Value
TRUE or FALSE
Filter an extension vector to those that participate in S3/S4 dispatch
Description
An extension participates in dispatch if its requirement is NA_character_
(in-development), if an S4 class with its name already exists, or if its
loaded package registers S3 dispatch methods for its marker class (see
providesDispatchMethods()). The last case lets an installed extension
package participate without defining its marker class statically; mizer
creates the class dynamically in defineExtensionClasses().
Usage
dispatchExtensions(extensions)
Arguments
extensions |
Named character vector of extensions. |
Value
A named character vector containing only the dispatch extensions, preserving order.
Measure distance between current and previous state in terms of RDI
Description
This function can be used in projectToSteady() to decide when sufficient
convergence to steady state has been achieved.
Usage
distanceMaxRelRDI(params, current, previous)
Arguments
params |
MizerParams |
current |
A named list with entries |
previous |
A named list with entries |
Value
The largest absolute relative change in rdi:
max(abs((current_rdi - previous_rdi) / previous_rdi)). If any entry of
previous_rdi is zero, the result can be infinite.
See Also
Other distance functions:
distanceSSLogN()
Measure distance between current and previous state in terms of fish abundances
Description
Calculates the sum squared difference between log(N) in current and previous
state. This function can be used in projectToSteady() to decide when
sufficient convergence to steady state has been achieved.
Usage
distanceSSLogN(params, current, previous)
Arguments
params |
MizerParams |
current |
A named list with entries |
previous |
A named list with entries |
Value
The sum of squares of the difference in the logs of the (nonzero)
fish abundances n, ignoring entries where either state has zero
abundance:
sum((log(current$n) - log(previous$n))^2)
See Also
Other distance functions:
distanceMaxRelRDI()
Length based double-sigmoid selectivity function
Description
A hump-shaped selectivity function with a sigmoidal rise and an independent
sigmoidal drop-off. This drop-off is what distinguishes this from the
function sigmoid_length() and it is intended to model the escape of large
individuals from the fishing gear.
Usage
double_sigmoid_length(w, l25, l50, l50_right, l25_right, species_params, ...)
Arguments
w |
Vector of sizes. |
l25 |
the length which gives a selectivity of 25%. |
l50 |
the length which gives a selectivity of 50%. |
l50_right |
the length which gives a selectivity of 50%. |
l25_right |
the length which gives a selectivity of 25%. |
species_params |
A list with the species params for the current species.
Used to get at the length-weight parameters |
... |
Unused |
Details
You would not usually call this function directly. Instead, set the sel_func
column in gear_params() to "double_sigmoid_length" and provide the
l25, l50, l50_right and l25_right values as additional columns.
setFishing() will then call this function automatically when calculating
the selectivity array.
The selectivity is obtained as the product of two sigmoidal curves, one
rising and one dropping. The sigmoidal rise is based on the two parameters
l25 and l50 which determine the length at which 25% and 50% of
the stock is selected respectively. The sigmoidal drop-off is based on the
two parameters l50_right and l25_right which determine the
length at which the selectivity curve has dropped back to 50% and 25%
respectively. The selectivity is given by the function
S(l) =
\frac{1}{1 + \exp\left(\log(3)\frac{l50 -l}{l50 - l25}\right)}\frac{1}{1 +
\exp\left(\log(3)\frac{l50_{right} -l}{l50_{right} -
l25_{right}}\right)}
As the size-based model is weight based, and this selectivity function is
length based, it uses the length-weight parameters a and b to convert
between length and weight.
l = \left(\frac{w}{a}\right)^{1/b}
Value
Vector of selectivities at the given sizes.
Requires l25 < l50 < l50_right < l25_right.
See Also
gear_params() for setting the selectivity parameters.
Other selectivity functions:
knife_edge(),
sigmoid_length(),
sigmoid_weight()
Examples
# Hump-shaped selectivity: rises from l25=10 to l50=15,
# then drops back to 50% at l50_right=40 and 25% at l25_right=50
sp <- list(a = 0.01, b = 3)
w <- c(1, 10, 100, 500, 1000)
double_sigmoid_length(w, l25 = 10, l50 = 15,
l50_right = 40, l25_right = 50,
species_params = sp)
Create empty MizerParams object of the right size
Description
An internal function. Sets up a valid MizerParams object with all the slots initialised and given dimension names, but with some slots left empty. This function is to be used by other functions to set up full parameter objects.
Usage
emptyParams(
species_params,
gear_params = data.frame(),
no_w = 100,
min_w = 0.001,
max_w = NA,
min_w_pp = 1e-12
)
Arguments
species_params |
A data frame of species-specific parameter values. |
gear_params |
A data frame with gear-specific parameter values. |
no_w |
The number of size bins in the consumer spectrum. |
min_w |
Sets the size of the eggs of all species for which this is not
given in the |
max_w |
The largest size of the consumer spectrum. By default this is
set to the largest |
min_w_pp |
The smallest size of the resource spectrum. |
Value
An empty but valid MizerParams object
Size grid
A size grid is created so that
the log-sizes are equally spaced. The spacing is chosen so that there will be
no_w fish size bins, with the smallest starting at min_w and the largest
starting at max_w. For the resource spectrum there is a larger set of
bins containing additional bins below
min_w, with the same log size. The number of extra bins is such that
min_w_pp comes to lie within the smallest bin.
Changes to species params
The species_params slot of the returned MizerParams object may differ
from the data frame supplied as argument to this function because
default values are set for missing parameters.
See Also
See newMultispeciesParams() for a function that fills
the slots left empty by this function.
Load (and optionally install) namespaces for all non-NA extensions
Description
For each extension whose requirement is not NA_character_, checks that the
package is installed and up-to-date, installs or upgrades via
pak::pkg_install() if install = TRUE, then calls loadNamespace().
Usage
ensureExtensionNamespaces(extensions, install = FALSE)
Arguments
extensions |
Named character vector of extensions. |
install |
Logical. If |
Value
Invisibly TRUE.
Expand the size grid
Description
This function expands the size grid in a MizerParams object to the desired
min and max size, preserving all existing species. The function is deprecated
because you can achieve the same more flexibly with adjustSizeGrid().
Usage
expandSizeGrid(params, ...)
## S3 method for class 'MizerParams'
expandSizeGrid(
params,
new_min_w = min(params@w),
new_max_w = max(params@w),
preserve_species = params@species_params$species,
...
)
Arguments
params |
A MizerParams object. |
... |
Additional arguments (currently unused). |
new_min_w |
The new minimum size in the grid. Defaults to the current minimum. |
new_max_w |
The new maximum size in the grid. Defaults to the current maximum. |
preserve_species |
A vector of species names for which all rate arrays should be copied over to the new params object rather than being re-calculated from the species parameters. If missing, all species are preserved. |
Value
A new MizerParams object with the updated size grid.
Extract the requirement view of an extension chain
Description
The @extensions slot may be stored either as a named character vector of
requirement strings (the legacy/unversioned form) or as a named list whose
entries are length-2 character vectors c(requirement = ..., version = ...).
This helper returns the requirement strings as a plain named character
vector, which is the form used for dispatch and suffix comparison.
Usage
extensionRequirements(ext)
Arguments
ext |
The contents of an |
Value
A named character vector of requirement strings.
Get the recorded version stamp for one extension on an object
Description
Get the recorded version stamp for one extension on an object
Usage
extensionVersion(params, name)
Arguments
params |
A |
name |
The extension identifier. |
Value
The recorded version string, or NA_character_ if none.
Extract the version stamps of an extension chain
Description
Returns the version of the extension package that last upgraded the object
for each extension, or NA_character_ where no stamp is recorded (including
the legacy character-vector form, which carries no versions).
Usage
extensionVersions(ext)
Arguments
ext |
The contents of an |
Value
A named character vector of version strings (NA where unknown).
Whether any extension recorded on an object needs upgrading
Description
Returns TRUE if, for any extension recorded in the object's @extensions
slot whose package is installed, the recorded version stamp is missing (NA)
or older than the installed version of that package. A missing stamp counts
as needing an upgrade so that objects created before extension-version
tracking are brought up to date (and stamped) on first use; this is safe
because runExtensionUpgrades() only calls an upgrade method if one is registered and
such methods are written to be idempotent.
Usage
extension_needs_upgrading(params)
Arguments
params |
A MizerParams object. |
Value
TRUE or FALSE.
Filter plotting data to the requested length limits
Description
Filter plotting data to the requested length limits
Usage
filter_plot_length_limits(plot_dat, llim)
Arguments
plot_dat |
A data frame of plotting data, possibly with an |
llim |
Numeric vector of length two giving the lower and upper length
limits. Use |
Value
plot_dat filtered to the length limits, or unchanged if it has no
l column.
Size spectra at end of simulation
Description
Size spectra at end of simulation
Usage
finalN(sim)
finalNResource(sim)
idxFinalT(sim)
Arguments
sim |
A MizerSim object |
Value
For finalN(): An ArraySpeciesBySize object (species x size)
holding the consumer number densities at the end of the simulation
For finalNResource(): A vector holding the resource number
densities at the end of the simulation for all size classes
For idxFinalT(): An integer giving the index for extracting the
results for the final time step
Examples
str(finalN(NS_sim))
# This could also be obtained using `N()` and `idxFinalT()`
identical(N(NS_sim)[idxFinalT(NS_sim), , ], finalN(NS_sim))
str(finalNResource(NS_sim))
idx <- idxFinalT(NS_sim)
idx
# This coincides with
length(getTimes(NS_sim))
# and corresponds to the final time
getTimes(NS_sim)[idx]
# We can use this index to extract the result at the final time
identical(N(NS_sim)[idx, , ], finalN(NS_sim))
identical(NResource(NS_sim)[idx, ], finalNResource(NS_sim))
Extract the final state from a simulation
Description
Returns the MizerParams object underlying the simulation with its initial
abundances set to the abundances at the last saved time step of the
simulation. This is a convenience wrapper around getParams() with no
time_range argument (the default).
Usage
finalParams(sim)
Arguments
sim |
A |
Value
A MizerParams object with initial values taken from the final time
step of the simulation.
See Also
Examples
sim <- project(NS_params, t_max = 20, effort = 0.5)
params_end <- finalParams(sim)
Assemble the flux matrix from growth, diffusion and recruitment rates
Description
Internal helper holding the arithmetic shared by getFlux.MizerParams and
getFlux.MizerSim. Keeping it separate lets the MizerSim method resolve the
rate functions once and reuse them across all saved time steps.
Usage
flux_from_rates(params, n, g, d, rdd, power = 0, flux_limiter = "none")
Arguments
params |
A valid |
n |
A matrix of species abundances (species x size). |
g |
Growth rate matrix (species x size), as from |
d |
Diffusion rate matrix (species x size), as from |
rdd |
Density-dependent reproduction rate vector (one per species), as
from |
power |
The flux at weight |
flux_limiter |
Advective-flux scheme: |
Value
A plain species x size matrix of fluxes (no mizer array class).
Units string for the flux returned by getFlux()
Description
Units string for the flux returned by getFlux()
Usage
flux_units(power)
Arguments
power |
The power of weight the flux was multiplied by. |
Value
A character string with the units of the flux.
Format an extension chain as a human-readable string
Description
Format an extension chain as a human-readable string
Usage
formatExtensionChain(extensions)
Arguments
extensions |
Named character vector of extensions. |
Value
A character string such as "mizerExtB -> mizerExtA", or
"<empty>" for a zero-length chain.
Gear parameters
Description
These functions allow you to get or set the gear parameters stored in
a MizerParams object. These are used by setFishing() to set up the
selectivity and catchability and thus together with the fishing effort
determine the fishing mortality.
Usage
gear_params(object)
gear_params(object) <- value
is.gear_params(x)
Arguments
object |
A MizerParams object, a MizerSim object or a data frame |
value |
A data frame with the new gear parameters. |
x |
An object to test with |
Details
The gear_params data has one row for each gear-species pair and one
column for each parameter that determines how that gear interacts with that
species. The columns are:
-
speciesThe name of the species -
gearThe name of the gear -
catchabilityA number specifying how strongly this gear selects this species. -
sel_funcThe name of the function that calculates the selectivity curve. One column for each selectivity parameter needed by the selectivity functions.
For the details see setFishing().
There can optionally also be a column yield_observed that allows you to
specify for each gear and species the total annual fisheries yield.
The fishing effort, which is also needed to determine the fishing mortality
exerted by a gear is not set via the gear_params data frame but is set
with initial_effort() or is specified when calling project().
If you change a gear parameter, this will be used to recalculate the
selectivity and catchability arrays by calling setFishing(),
unless you have previously set these by hand.
gear_params<- automatically sets the row names to contain the species name
and the gear name, separated by a comma and a space. The last example below
illustrates how this facilitates changing an individual gear parameter.
Value
Data frame with gear parameters
is.gear_params() returns TRUE if x is a gear_params object,
FALSE otherwise.
See Also
Other functions for setting parameters:
setExtDiffusion(),
setExtEncounter(),
setExtMort(),
setFishing(),
setInteraction(),
setMaxIntakeRate(),
setMetabolicRate(),
setParams(),
setPredKernel(),
setReproduction(),
setSearchVolume(),
species_params(),
use_predation_diffusion()
Examples
params <- NS_params
# gears set up in example
gear_params(params)
# setting totally different gears
gear_params(params) <- data.frame(
gear = c("gear1", "gear2", "gear1"),
species = c("Cod", "Cod", "Haddock"),
catchability = c(0.5, 2, 1),
sel_func = c("sigmoid_weight", "knife_edge", "sigmoid_weight"),
sigmoidal_weight = c(1000, NA, 800),
sigmoidal_sigma = c(100, NA, 100),
knife_edge_size = c(NA, 1000, NA)
)
gear_params(params)
# changing an individual entry
gear_params(params)["Cod, gear1", "catchability"] <- 0.8
Calculate the total biomass of each species within a size range at each time step.
Description
Calculates the total biomass through time within user defined size limits.
The default option is to use the size range starting at the size specified
by the biomass_cutoff species parameter, if it is set, or else the full
size range of each species. You can specify minimum
and maximum weight or length range for the species. Lengths take precedence
over weights (i.e. if both min_l and min_w are supplied, only min_l will be
used).
Usage
getBiomass(object, use_cutoff = FALSE, ...)
Arguments
object |
An object of class |
use_cutoff |
If TRUE, the |
... |
Arguments passed on to
|
Details
When no size range arguments are provided, the function checks if the
biomass_cutoff column exists in the species parameters. If it does,
those values are used as the minimum weight for each species. For species
with NA values in biomass_cutoff, the default minimum weight (smallest
weight in the model) is used.
Value
If called with a MizerParams object, a named vector with the biomass
in grams for each species in the model. If called with a MizerSim object,
an ArrayTimeBySpecies object (time x species) containing the biomass in
grams at each time step for all species.
See Also
Other summary functions:
getDiet(),
getGrowthCurves(),
getN(),
getSSB(),
getTrophicLevel(),
getTrophicLevelBySpecies(),
getYield(),
getYieldGear()
Examples
biomass <- getBiomass(NS_sim)
biomass["1972", "Herring"]
biomass <- getBiomass(NS_sim, min_w = 10, max_w = 1000)
biomass["1972", "Herring"]
# If species_params contains a `biomass_cutoff`` column, it can be used
# as the minimum weight when use_cutoff = TRUE
species_params(NS_sim@params)$biomass_cutoff <- 10
biomass <- getBiomass(NS_sim, use_cutoff = TRUE) # Uses biomass_cutoff as min_w
biomass["1972", "Herring"]
Calculate the slope of the community abundance
Description
Calculates the slope of the community abundance by performing a linear regression on the logged total numerical abundance at weight and logged weights (natural logs, not log to base 10, are used). You can specify minimum and maximum weight or length range for the species. Lengths take precedence over weights (i.e. if both min_l and min_w are supplied, only min_l will be used). You can also specify the species to be used in the calculation.
Usage
getCommunitySlope(object, species = NULL, biomass = TRUE, ...)
Arguments
object |
A MizerSim or MizerParams object |
species |
The species to be selected. Optional. By default all target species are selected. A vector of species names, or a numeric vector with the species indices, or a logical vector indicating for each species whether it is to be selected (TRUE) or not. |
biomass |
Boolean. If TRUE (default), the abundance is based on biomass, if FALSE the abundance is based on numbers. |
... |
Arguments passed on to
|
Value
A data.frame with columns slope, intercept and the coefficient of
determination R^2 (and a time step column when called with a MizerSim
object).
See Also
Other functions for calculating indicators:
getMeanMaxWeight(),
getMeanWeight(),
getProportionOfLargeFish()
Examples
# Slope based on biomass, using all species and sizes
slope_biomass <- getCommunitySlope(NS_sim)
slope_biomass[1, ] # in 1976
slope_biomass[idxFinalT(NS_sim), ] # in 2010
# Slope based on numbers, using all species and sizes
slope_numbers <- getCommunitySlope(NS_sim, biomass = FALSE)
slope_numbers[1, ] # in 1976
# Slope based on biomass, using all species and sizes between 10g and 1000g
slope_biomass <- getCommunitySlope(NS_sim, min_w = 10, max_w = 1000)
slope_biomass[1, ] # in 1976
# Slope based on biomass, using only demersal species and
# sizes between 10g and 1000g
dem_species <- c("Dab","Whiting", "Sole", "Gurnard", "Plaice",
"Haddock", "Cod", "Saithe")
slope_biomass <- getCommunitySlope(NS_sim, species = dem_species,
min_w = 10, max_w = 1000)
slope_biomass[1, ] # in 1976
getCommunitySlope(NS_params)
Get critical feeding level
Description
The critical feeding level is the feeding level at which the food intake is just high enough to cover the metabolic costs, with nothing left over for growth or reproduction.
Usage
getCriticalFeedingLevel(params)
Arguments
params |
A MizerParams object |
Value
An ArraySpeciesBySize object (species x size) with the critical feeding level
Examples
str(getFeedingLevel(NS_params))
Get diet of predator at size, resolved by prey species
Description
Calculates the rate at which a predator of a particular species and size consumes biomass of each prey species, resource, and other components of the ecosystem. Returns either the rates in grams/year or the proportion of the total consumption rate.
Usage
getDiet(object, proportion = TRUE, ...)
Arguments
object |
A MizerParams or MizerSim object. |
proportion |
If TRUE (default) the function returns the diet as a proportion of the total consumption rate. If FALSE it returns the consumption rate in grams per year. |
... |
Additional arguments that depend on the class of For a MizerParams object:
For a MizerSim object:
|
Details
The rates D_{ij}(w) at which a predator of species i
and size w consumes biomass from prey species j are
calculated from the predation kernel \phi_i(w, w_p),
the search volume \gamma_i(w), the feeding level f_i(w), the
species interaction matrix \theta_{ij} and the prey abundance density
N_j(w_p):
D_{ij}(w, w_p) = (1-f_i(w)) \gamma_i(w) \theta_{ij}
\int N_j(w_p) \phi_i(w, w_p) w_p dw_p.
The prey index j runs over all species and the resource.
Extra columns are added for the external encounter rate and for any extra
ecosystem components in your model for which you have defined an encounter
rate function. These encounter rates are multiplied by 1-f_i(w) to give
the rate of consumption of biomass from these extra components.
This function performs the same integration as getEncounter() but does not
aggregate over prey species, and multiplies by 1-f_i(w) to get the
consumed biomass rather than the available biomass. Outside the range of
sizes for a predator species the returned rate is zero.
Value
-
MizerParams: An array (predator species x predator size x (prey species + resource + other components)). Dimnames are "predator", "w", and "prey". -
MizerSim: A four-dimensional array (time x predator species x predator size x prey) with the diet at each selected saved time step. Ifdrop = TRUEthen dimensions of length 1 are removed.
See Also
Other summary functions:
getBiomass(),
getGrowthCurves(),
getN(),
getSSB(),
getTrophicLevel(),
getTrophicLevelBySpecies(),
getYield(),
getYieldGear()
Examples
diet <- getDiet(NS_params)
str(diet)
# For a MizerSim the diet is returned at each saved time step
sim <- project(NS_params, t_max = 20, effort = 0.5)
# Diet at the saved time steps over years 15 - 20
diet <- getDiet(sim, time_range = c(15, 20))
str(diet)
Get diffusion rate from predation
Description
Calculates the diffusion rate D_i(w) (grams^2/year) for each species.
This diffusion rate has two components:
The diffusion due due to the variability in prey sizes. This is the diffusion term from the jump-growth equation.
Any externally specified diffusion, which is added via
setExtDiffusion()
Usage
getDiffusion(object, ...)
Arguments
object |
A MizerParams or MizerSim object. |
... |
Additional arguments that depend on the class of For a MizerParams object:
For a MizerSim object:
|
Details
The diffusion due due to the variability in prey sizes
is determined by summing over all prey
species and the resource spectrum and then integrating over all prey sizes
w_p, weighted by predation kernel \phi(w,w_p):
d_i(w) = (1-f_i(w))(\alpha_i(1-\psi_i(w)))^2\gamma_i(w) \int
\left( \theta_{ip} N_R(w_p) + \sum_{j} \theta_{ij} N_j(w_p) \right)
\phi_i(w,w_p) w_p^2 \, dw_p.
Here N_j(w) is the abundance density of species j and
N_R(w) is the abundance density of resource.
The overall prefactor \gamma_i(w) determines the predation power of the
predator. It could be interpreted as a search volume and is set with the
setSearchVolume() function. The predation kernel
\phi(w,w_p) is set with the setPredKernel() function. The
species interaction matrix \theta_{ij} is set with setInteraction()
and the resource interaction vector \theta_{ip} is taken from the
interaction_resource column in species_params().
f(w) is the feeding level calculated with
getFeedingLevel(). \psi(w) is the proportion of the available energy
that is invested in reproduction instead of growth, obtained with psi().
The diffusion integral is normally evaluated efficiently with a fast Fourier
transform, which assumes that the predation kernel depends only on the ratio
of predator to prey size. If a custom predation kernel that depends on
predator and prey size separately has been set with setPredKernel(), the
integral is instead evaluated by direct summation over the full predation
kernel, as in getEncounter().
Value
-
MizerParams: AnArraySpeciesBySizeobject (predator species x predator size) with the diffusion rates. -
MizerSim: AnArrayTimeBySpeciesBySizeobject (time step x predator species x predator size) with the diffusion rates at every time step. Ifdrop = TRUEthen dimensions of length 1 will be removed.
References
Datta, S., Delius, G. W. and Law, R. (2010). A jump-growth model for predator-prey dynamics: derivation and application to marine ecosystems. Bulletin of Mathematical Biology, 72(6):1361–1382
See Also
Other rate functions:
getEGrowth(),
getERepro(),
getEReproAndGrowth(),
getEncounter(),
getFMort(),
getFMortGear(),
getFeedingLevel(),
getFlux(),
getFluxGradient(),
getMort(),
getPredMort(),
getPredRate(),
getRDD(),
getRDI(),
getRates(),
getResourceMort()
Get energy rate available for growth
Description
Calculates the energy rate g_i(w) (grams/year) available by species and
size for growth after metabolism, movement and reproduction have been
accounted for.
Usage
getEGrowth(object, ...)
Arguments
object |
A MizerParams or MizerSim object. |
... |
Additional arguments that depend on the class of For a MizerParams object:
For a MizerSim object:
|
Details
The growth rate is calculated as the difference between the energy available
for reproduction and growth (obtainable with getEReproAndGrowth()) and
the energy used for reproduction (obtainable with getERepro()), but is
set to 0 if the result would be negative.
Value
-
MizerParams: AnArraySpeciesBySizeobject (species x size) with the somatic growth rates (grams/year). -
MizerSim: AnArrayTimeBySpeciesBySizeobject (time step x species x size) with the growth rates at every time step. Ifdrop = TRUEthen dimensions of length 1 will be removed.
Your own growth rate function
By default getEGrowth() calls mizerEGrowth(). However you can
replace this with your own alternative growth rate function. If
your function is called "myEGrowth" then you register it in a MizerParams
object params with
params <- setRateFunction(params, "EGrowth", "myEGrowth")
Your function will then be called instead of mizerEGrowth(), with the
same arguments.
See Also
getERepro(), getEReproAndGrowth()
Other rate functions:
getDiffusion(),
getERepro(),
getEReproAndGrowth(),
getEncounter(),
getFMort(),
getFMortGear(),
getFeedingLevel(),
getFlux(),
getFluxGradient(),
getMort(),
getPredMort(),
getPredRate(),
getRDD(),
getRDI(),
getRates(),
getResourceMort()
Examples
params <- NS_params
# Project with constant fishing effort for all gears for 20 time steps
sim <- project(params, t_max = 20, effort = 0.5)
# Get the energy at a particular time step
growth <- getEGrowth(params, n = N(sim)[15, , ], n_pp = NResource(sim)[15, ], t = 15)
# Growth rate at this time for Sprat of size 2g
growth["Sprat", "2"]
Get energy rate available for reproduction
Description
Calculates the energy rate (grams/year) available for reproduction after growth and metabolism have been accounted for.
Usage
getERepro(object, ...)
Arguments
object |
A MizerParams or MizerSim object. |
... |
Additional arguments that depend on the class of For a MizerParams object:
For a MizerSim object:
|
Value
-
MizerParams: AnArraySpeciesBySizeobject (species x size) holding\psi_i(w)\max(0, E_{r.i}(w))where
E_{r.i}(w)is the rate at which energy becomes available for growth and reproduction, calculated withgetEReproAndGrowth(), and\psi_i(w)is the proportion of this energy that is used for reproduction. Negative values ofE_{r.i}(w)are clipped to 0 before multiplying by\psi_i(w). This proportion is taken from theparamsobject and is set withsetReproduction(). -
MizerSim: AnArrayTimeBySpeciesBySizeobject (time step x species x size) with the energy for reproduction at every time step. Ifdrop = TRUEthen dimensions of length 1 will be removed.
Your own reproduction rate function
By default getERepro() calls mizerERepro(). However you can
replace this with your own alternative reproduction rate function. If
your function is called "myERepro" then you register it in a MizerParams
object params with
params <- setRateFunction(params, "ERepro", "myERepro")
Your function will then be called instead of mizerERepro(), with the
same arguments.
See Also
Other rate functions:
getDiffusion(),
getEGrowth(),
getEReproAndGrowth(),
getEncounter(),
getFMort(),
getFMortGear(),
getFeedingLevel(),
getFlux(),
getFluxGradient(),
getMort(),
getPredMort(),
getPredRate(),
getRDD(),
getRDI(),
getRates(),
getResourceMort()
Examples
params <- NS_params
# Project with constant fishing effort for all gears for 20 time steps
sim <- project(params, t_max = 20, effort = 0.5)
# Get the rate at a particular time step
erepro <- getERepro(params, n = N(sim)[15, , ], n_pp = NResource(sim)[15, ], t = 15)
# Rate at this time for Sprat of size 2g
erepro["Sprat", "2"]
Get energy rate available for reproduction and growth
Description
Calculates the energy rate E_{r.i}(w) (grams/year) available for
reproduction and growth after metabolism and movement have been accounted
for.
Usage
getEReproAndGrowth(object, ...)
Arguments
object |
A MizerParams or MizerSim object. |
... |
Additional arguments that depend on the class of For a MizerParams object:
For a MizerSim object:
|
Value
-
MizerParams: AnArraySpeciesBySizeobject (species x size) with the energy rateE_{r.i}(w)available for growth and reproduction (grams/year). -
MizerSim: AnArrayTimeBySpeciesBySizeobject (time step x species x size) with the energy rate at every time step. Ifdrop = TRUEthen dimensions of length 1 will be removed.
Your own energy rate function
By default getEReproAndGrowth() calls mizerEReproAndGrowth(). However you
can replace this with your own alternative energy rate function. If
your function is called "myEReproAndGrowth" then you register it in a
MizerParams object params with
params <- setRateFunction(params, "EReproAndGrowth", "myEReproAndGrowth")
Your function will then be called instead of mizerEReproAndGrowth(), with
the same arguments.
See Also
The part of this energy rate that is invested into growth is
calculated with getEGrowth() and the part that is invested into
reproduction is calculated with getERepro().
Other rate functions:
getDiffusion(),
getEGrowth(),
getERepro(),
getEncounter(),
getFMort(),
getFMortGear(),
getFeedingLevel(),
getFlux(),
getFluxGradient(),
getMort(),
getPredMort(),
getPredRate(),
getRDD(),
getRDI(),
getRates(),
getResourceMort()
Examples
params <- NS_params
# Project with constant fishing effort for all gears for 20 time steps
sim <- project(params, t_max = 20, effort = 0.5)
# Get the energy at a particular time step
e <- getEReproAndGrowth(params, n = N(sim)[15, , ],
n_pp = NResource(sim)[15, ], t = 15)
# Rate at this time for Sprat of size 2g
e["Sprat", "2"]
Alias for getERepro()
Description
An alias provided for backward compatibility with mizer version <= 1.0
Usage
getESpawning(object, ...)
Arguments
object |
A MizerParams or MizerSim object. |
... |
Additional arguments that depend on the class of For a MizerParams object:
For a MizerSim object:
|
Value
-
MizerParams: AnArraySpeciesBySizeobject (species x size) holding\psi_i(w)\max(0, E_{r.i}(w))where
E_{r.i}(w)is the rate at which energy becomes available for growth and reproduction, calculated withgetEReproAndGrowth(), and\psi_i(w)is the proportion of this energy that is used for reproduction. Negative values ofE_{r.i}(w)are clipped to 0 before multiplying by\psi_i(w). This proportion is taken from theparamsobject and is set withsetReproduction(). -
MizerSim: AnArrayTimeBySpeciesBySizeobject (time step x species x size) with the energy for reproduction at every time step. Ifdrop = TRUEthen dimensions of length 1 will be removed.
Your own reproduction rate function
By default getERepro() calls mizerERepro(). However you can
replace this with your own alternative reproduction rate function. If
your function is called "myERepro" then you register it in a MizerParams
object params with
params <- setRateFunction(params, "ERepro", "myERepro")
Your function will then be called instead of mizerERepro(), with the
same arguments.
See Also
Other rate functions:
getDiffusion(),
getEGrowth(),
getEReproAndGrowth(),
getEncounter(),
getFMort(),
getFMortGear(),
getFeedingLevel(),
getFlux(),
getFluxGradient(),
getMort(),
getPredMort(),
getPredRate(),
getRDD(),
getRDI(),
getRates(),
getResourceMort()
Examples
params <- NS_params
# Project with constant fishing effort for all gears for 20 time steps
sim <- project(params, t_max = 20, effort = 0.5)
# Get the rate at a particular time step
erepro <- getERepro(params, n = N(sim)[15, , ], n_pp = NResource(sim)[15, ], t = 15)
# Rate at this time for Sprat of size 2g
erepro["Sprat", "2"]
Fishing effort used in simulation
Description
Note that the array returned may not be exactly the same as the effort
argument that was passed in to project(). This is because only the saved
effort is stored (the frequency of saving is determined by the argument
t_save).
Usage
getEffort(sim)
Arguments
sim |
A MizerSim object |
Value
An array (time x gear) that contains the fishing effort by time and gear.
Examples
str(getEffort(NS_sim))
Get encounter rate
Description
Returns the rate at which a predator of species i and
weight w encounters food (grams/year).
Usage
getEncounter(object, ...)
Arguments
object |
A MizerParams or MizerSim object. |
... |
Additional arguments that depend on the class of For a MizerParams object:
For a MizerSim object:
|
Value
-
MizerParams: AnArraySpeciesBySizeobject (predator species x predator size) with the encounter rates. -
MizerSim: AnArrayTimeBySpeciesBySizeobject (time step x predator species x predator size) with the encounter rates at every time step. Ifdrop = TRUEthen dimensions of length 1 will be removed.
Predation encounter
The encounter rate E_i(w) at which a predator of species i
and weight w encounters food has contributions from the encounter of
fish prey and of resource. This is determined by summing over all prey
species and the resource spectrum and then integrating over all prey sizes
w_p, weighted by predation kernel \phi(w,w_p):
E_i(w) = \gamma_i(w) \int
\left( \theta_{ip} N_R(w_p) + \sum_{j} \theta_{ij} N_j(w_p) \right)
\phi_i(w,w_p) w_p \, dw_p.
Here N_j(w) is the abundance density of species j and
N_R(w) is the abundance density of resource.
The overall prefactor \gamma_i(w) determines the predation power of the
predator. It could be interpreted as a search volume and is set with the
setSearchVolume() function. The predation kernel
\phi(w,w_p) is set with the setPredKernel() function. The
species interaction matrix \theta_{ij} is set with setInteraction()
and the resource interaction vector \theta_{ip} is taken from the
interaction_resource column in params@species_params.
Details
The encounter rate is multiplied by 1-f_0 to obtain the consumption
rate, where f_0 is the feeding level calculated with
getFeedingLevel(). This is used by the project() function for performing
simulations.
The function returns values also for sizes outside the size-range of the species. These values should not be used, as they are meaningless.
If your model contains additional components that you added with
setComponent() and for which you specified an encounter_fun function then
the encounters of these components will be included in the returned value.
Extension hook
projectEncounter() is the S3 generic used by extension-aware projections.
Extension packages can add methods for their marker classes and call
NextMethod() to compose encounter-rate changes. The MizerParams method
contains the standard mizer calculation and is also exported as
mizerEncounter() for compatibility.
Your own encounter function
By default getEncounter() calls mizerEncounter() on models without
extensions. However you can replace this with your own alternative encounter
function. If your function is called "myEncounter" then you register it in
a MizerParams object params with
params <- setRateFunction(params, "Encounter", "myEncounter")
Your function will then be called instead of mizerEncounter(), with the
same arguments.
See Also
Other rate functions:
getDiffusion(),
getEGrowth(),
getERepro(),
getEReproAndGrowth(),
getFMort(),
getFMortGear(),
getFeedingLevel(),
getFlux(),
getFluxGradient(),
getMort(),
getPredMort(),
getPredRate(),
getRDD(),
getRDI(),
getRates(),
getResourceMort()
Examples
encounter <- getEncounter(NS_params)
str(encounter)
Get the total fishing mortality rate from all fishing gears by time, species and size.
Description
Calculates the total fishing mortality (in units 1/year) from all gears by
species and size and possibly time. See setFishing() for details of
how fishing gears are set up.
Usage
getFMort(object, ...)
Arguments
object |
A MizerParams or MizerSim object. |
... |
Additional arguments that depend on the class of For a MizerParams object:
For a MizerSim object:
|
Details
The total fishing mortality is just the sum of the fishing mortalities
imposed by each gear, F_i(w)=\sum_g F_{g,i,w}.
The fishing mortality for each gear is obtained as catchability x
selectivity x effort.
Value
-
MizerParamswith vector effort: AnArraySpeciesBySizeobject (species x size) with the fishing mortality rates. -
MizerParamswith time-dimensioned effort orMizerSim: AnArrayTimeBySpeciesBySizeobject (time x species x size).
The effort argument is only used if a MizerParams object is
passed in. The effort argument can be a two dimensional array (time x
gear), a vector of length equal to the number of gears (each gear has a
different effort that is constant in time), or a single numeric value (each
gear has the same effort that is constant in time). The order of gears in the
effort argument must be the same as in the MizerParams
object.
If the object argument is of class MizerSim then the effort slot of
the MizerSim object is used and the effort argument is not
used.
Your own fishing mortality function
By default getFMort() calls mizerFMort(). However you can
replace this with your own alternative fishing mortality function. If
your function is called "myFMort" then you register it in a MizerParams
object params with
params <- setRateFunction(params, "FMort", "myFMort")
Your function will then be called instead of mizerFMort(), with the
same arguments.
See Also
Other rate functions:
getDiffusion(),
getEGrowth(),
getERepro(),
getEReproAndGrowth(),
getEncounter(),
getFMortGear(),
getFeedingLevel(),
getFlux(),
getFluxGradient(),
getMort(),
getPredMort(),
getPredRate(),
getRDD(),
getRDI(),
getRates(),
getResourceMort()
Examples
params <- NS_params
# Get the total fishing mortality in the initial state
F <- getFMort(params, effort = 1)
str(F)
# Get the initial total fishing mortality when effort is different
# between the four gears:
F <- getFMort(params, effort = c(0.5,1,1.5,0.75))
# Get the total fishing mortality when effort is different
# between the four gears and changes with time:
effort <- array(NA, dim = c(20,4))
effort[, 1] <- seq(from = 0, to = 1, length = 20)
effort[, 2] <- seq(from = 1, to = 0.5, length = 20)
effort[, 3] <- seq(from = 1, to = 2, length = 20)
effort[, 4] <- seq(from = 2, to = 1, length = 20)
F <- getFMort(params, effort = effort)
str(F)
# Get the total fishing mortality using the effort already held in a
# MizerSim object.
sim <- project(params, t_max = 20, effort = 0.5)
F <- getFMort(sim)
F <- getFMort(sim, time_range = c(10, 20))
Get the fishing mortality by time, gear, species and size
Description
Calculates the fishing mortality rate F_{g,i,w} by gear, species and
size and possibly time (in units 1/year).
Usage
getFMortGear(object, ...)
Arguments
object |
A MizerParams or MizerSim object. |
... |
Additional arguments that depend on the class of For a MizerParams object:
For a MizerSim object:
|
Value
An array. If the effort argument has a time dimension, or a
MizerSim is passed in, the output array has four dimensions (time x
gear x species x size). If the effort argument does not have a time
dimension (i.e. it is a vector or a single numeric), the output array has
three dimensions (gear x species x size).
Note
Here: fishing mortality = catchability x selectivity x effort.
The effort argument is only used if a MizerParams object is
passed in. The effort argument can be a two dimensional array (time x
gear), a vector of length equal to the number of gears (each gear has a
different effort that is constant in time), or a single numeric value (each
gear has the same effort that is constant in time). The order of gears in the
effort argument must be the same the same as in the MizerParams
object. If the effort argument is not supplied, its value is taken
from the @initial_effort slot in the params object.
If the object argument is of class MizerSim then the effort slot of
the MizerSim object is used and the effort argument is not
used.
See Also
Other rate functions:
getDiffusion(),
getEGrowth(),
getERepro(),
getEReproAndGrowth(),
getEncounter(),
getFMort(),
getFeedingLevel(),
getFlux(),
getFluxGradient(),
getMort(),
getPredMort(),
getPredRate(),
getRDD(),
getRDI(),
getRates(),
getResourceMort()
Examples
params <-NS_params
# Get the fishing mortality in initial state
F <- getFMortGear(params, effort = 1)
str(F)
# Get the initial fishing mortality when effort is different
# between the four gears:
F <- getFMortGear(params, effort = c(0.5, 1, 1.5, 0.75))
# Get the fishing mortality when effort is different
# between the four gears and changes with time:
effort <- array(NA, dim = c(20, 4))
effort[, 1] <- seq(from=0, to = 1, length = 20)
effort[, 2] <- seq(from=1, to = 0.5, length = 20)
effort[, 3] <- seq(from=1, to = 2, length = 20)
effort[, 4] <- seq(from=2, to = 1, length = 20)
F <- getFMortGear(params, effort = effort)
str(F)
# Get the fishing mortality using the effort already held in a MizerSim object.
sim <- project(params, t_max = 20, effort = 0.5)
F <- getFMortGear(sim)
F <- getFMortGear(sim, time_range = c(10, 20))
Get feeding level
Description
Returns the feeding level.
By default this function uses mizerFeedingLevel() to calculate
the feeding level, but this can be overruled via setRateFunction().
Usage
getFeedingLevel(object, ...)
Arguments
object |
A MizerParams or MizerSim object. |
... |
Additional arguments that depend on the class of For a MizerParams object:
For a MizerSim object:
|
Value
-
MizerParams: AnArraySpeciesBySizeobject (predator species x predator size) with the feeding level. -
MizerSim: AnArrayTimeBySpeciesBySizeobject (time step x predator species x predator size) with the feeding level at every time step. Ifdrop = TRUEthen dimensions of length 1 will be removed.
Feeding level
The feeding level f_i(w) is the
proportion of its maximum intake rate at which the predator is actually
taking in fish. It is calculated from the encounter rate E_i and the
maximum intake rate h_i(w) as
f_i(w) = \frac{E_i(w)}{E_i(w)+h_i(w)}.
The encounter rate E_i is passed as an argument or calculated with
getEncounter(). The maximum intake rate h_i(w) is
taken from the params object, and is set with
setMaxIntakeRate().
As a consequence of the above expression for the feeding level,
1-f_i(w) is the proportion of the food available to it that the
predator actually consumes.
Your own feeding level function
By default getFeedingLevel() calls mizerFeedingLevel(). However you can
replace this with your own alternative feeding level function. If
your function is called "myFeedingLevel" then you register it in a MizerParams
object params with
params <- setRateFunction(params, "FeedingLevel", "myFeedingLevel")
Your function will then be called instead of mizerFeedingLevel(), with the
same arguments.
See Also
Other rate functions:
getDiffusion(),
getEGrowth(),
getERepro(),
getEReproAndGrowth(),
getEncounter(),
getFMort(),
getFMortGear(),
getFlux(),
getFluxGradient(),
getMort(),
getPredMort(),
getPredRate(),
getRDD(),
getRDI(),
getRates(),
getResourceMort()
Examples
params <- NS_params
# Get initial feeding level
fl <- getFeedingLevel(params)
# Project with constant fishing effort for all gears for 20 time steps
sim <- project(params, t_max = 20, effort = 0.5)
# Get the feeding level at all saved time steps
fl <- getFeedingLevel(sim)
# Get the feeding level for years 15 - 20
fl <- getFeedingLevel(sim, time_range = c(15, 20))
Get flux into size bins
Description
Calculates the flux J_i(w) (numbers/year) entering each size class
from the one below it. This is composed of an advective flux from somatic
growth and a diffusive flux from the redistribution of individuals.
Usage
getFlux(object, ..., power = 0)
Arguments
object |
A MizerParams or MizerSim object. |
... |
Additional arguments that depend on the class of For a MizerParams object:
For a MizerSim object:
|
power |
The flux at weight |
Details
At the recruitment size, the flux is simply the recruitment rate
R_{dd,i} (see getRDD()). For sizes below the recruitment size
the flux is zero.
The flux at weight w is multiplied by w raised to the power
given by the power argument, similar to the power argument of
plotSpectra(). The default power = 0 returns the flux of individuals
(numbers/year). With power = 1 the result is the flux of biomass
(grams/year).
Value
-
MizerParams: AnArraySpeciesBySizeobject (species x size) with the flux entering each size class. The units arenumbers/yearwhenpower = 0andg^power/yearotherwise. -
MizerSim: AnArrayTimeBySpeciesBySizeobject (time step x species x size) with the flux at every time step. Ifdrop = TRUEthen dimensions of length 1 will be removed.
See Also
Other rate functions:
getDiffusion(),
getEGrowth(),
getERepro(),
getEReproAndGrowth(),
getEncounter(),
getFMort(),
getFMortGear(),
getFeedingLevel(),
getFluxGradient(),
getMort(),
getPredMort(),
getPredRate(),
getRDD(),
getRDI(),
getRates(),
getResourceMort()
Examples
params <- NS_params
# Project with constant fishing effort for all gears for 20 time steps
sim <- project(params, t_max = 20, effort = 0.5)
# Get the flux at a particular time step
flux <- getFlux(params, n = N(sim)[15, , ], n_pp = NResource(sim)[15, ], t = 15)
# Flux for Sprat of size 2g
flux["Sprat", "2"]
Get flux gradient
Description
Calculates the flux divergence (J_{j+1} - J_j)/\Delta w_j that
appears as the second term in the discretised size-spectrum transport
equation
\frac{\partial N_j}{\partial t} + \frac{J_{j+1} - J_j}{\Delta w_j}
= -\mu_j N_j.
The bin-boundary fluxes J_j are obtained from getFlux(), which
uses the advective-flux scheme stored in the flux entry of the
second_order_w slot of params. The flux leaving
the largest size class through the upper boundary (J_{K+1}) is
evaluated with the same scheme using the boundary condition
N_{K+1} = 0.
Usage
getFluxGradient(object, ...)
Arguments
object |
A MizerParams or MizerSim object. |
... |
Additional arguments that depend on the class of For a MizerParams object:
For a MizerSim object:
|
Value
-
MizerParams: AnArraySpeciesBySizeobject (species x size) giving the flux divergence in each size bin, in units ofg^{-1} \, \text{year}^{-1}. -
MizerSim: AnArrayTimeBySpeciesBySizeobject (time step x species x size) with the flux divergence at every saved time step. Ifdrop = TRUEthen dimensions of length 1 will be removed.
See Also
Other rate functions:
getDiffusion(),
getEGrowth(),
getERepro(),
getEReproAndGrowth(),
getEncounter(),
getFMort(),
getFMortGear(),
getFeedingLevel(),
getFlux(),
getMort(),
getPredMort(),
getPredRate(),
getRDD(),
getRDI(),
getRates(),
getResourceMort()
Examples
params <- NS_params
fg <- getFluxGradient(params)
sim <- project(params, t_max = 5)
fg_sim <- getFluxGradient(sim)
Get growth curves giving weight as a function of age
Description
Get growth curves giving weight as a function of age
Usage
getGrowthCurves(object, species = NULL, max_age = 20, percentage = FALSE)
Arguments
object |
MizerSim or MizerParams object. If given a MizerSim object, uses the growth rates at the final time of a simulation to calculate the size at age. If given a MizerParams object, uses the initial growth rates instead. |
species |
The species to be selected. Optional. By default all target species are selected. A vector of species names, or a numeric vector with the species indices, or a logical vector indicating for each species whether it is to be selected (TRUE) or not. |
max_age |
The age up to which to run the growth curve. Default is 20. |
percentage |
Boolean value. If TRUE, the size is given as a percentage of the maximal size. |
Value
An array (species x age) containing the weight in grams.
See Also
Other summary functions:
getBiomass(),
getDiet(),
getN(),
getSSB(),
getTrophicLevel(),
getTrophicLevelBySpecies(),
getYield(),
getYieldGear()
Examples
growth_curves <- getGrowthCurves(NS_params, species = c("Cod", "Haddock"))
str(growth_curves)
library(ggplot2)
ggplot(melt(growth_curves)) +
geom_line(aes(Age, value)) +
facet_wrap(~ Species, scales = "free") +
ylab("Size[g]") + xlab("Age[years]")
Deprecated function to get interaction matrix
Description
You should now use interaction_matrix() instead.
Usage
getInteraction(params)
Arguments
params |
A MizerParams object |
Alias for getPredMort()
Description
An alias provided for backward compatibility with mizer version <= 1.0
Usage
getM2(object, ...)
Arguments
object |
A MizerParams or MizerSim object. |
... |
Additional arguments that depend on the class of For a MizerParams object:
For a MizerSim object:
|
Value
-
MizerParams: AnArraySpeciesBySizeobject (prey species x prey size) with the predation mortality rates. -
MizerSim: AnArrayTimeBySpeciesBySizeobject (time step x prey species x prey size) with the predation mortality at every time step. Ifdrop = TRUEthen dimensions of length 1 will be removed.
Your own predation mortality function
By default getPredMort() calls mizerPredMort(). However you can
replace this with your own alternative predation mortality function. If
your function is called "myPredMort" then you register it in a MizerParams
object params with
params <- setRateFunction(params, "PredMort", "myPredMort")
Your function will then be called instead of mizerPredMort(), with the
same arguments.
See Also
Other rate functions:
getDiffusion(),
getEGrowth(),
getERepro(),
getEReproAndGrowth(),
getEncounter(),
getFMort(),
getFMortGear(),
getFeedingLevel(),
getFlux(),
getFluxGradient(),
getMort(),
getPredRate(),
getRDD(),
getRDI(),
getRates(),
getResourceMort()
Examples
params <- NS_params
# Predation mortality in initial state
M2 <- getPredMort(params)
str(M2)
# With constant fishing effort for all gears for 20 time steps
sim <- project(params, t_max = 20, effort = 0.5)
# Get predation mortality at one time step
M2 <- getPredMort(params, n = N(sim)[15, , ], n_pp = NResource(sim)[15, ])
# Get predation mortality at all saved time steps
M2 <- getPredMort(sim)
str(M2)
# Get predation mortality over the years 15 - 20
M2 <- getPredMort(sim, time_range = c(15, 20))
Alias for getResourceMort()
Description
An alias provided for backward compatibility with mizer version <= 1.0
Usage
getM2Background(
params,
n = initialN(params),
n_pp = initialNResource(params),
n_other = initialNOther(params),
t = 0,
...
)
Arguments
params |
A MizerParams object |
n |
A matrix of species abundances (species x size). |
n_pp |
A vector of the resource abundance by size |
n_other |
A list of abundances for other dynamical components of the ecosystem |
t |
The time for which to do the calculation (Not used by standard mizer rate functions but useful for extensions with time-dependent parameters.) |
... |
Unused |
Value
A vector of mortality rate by resource size.
Your own resource mortality function
By default getResourceMort() calls mizerResourceMort(). However you can
replace this with your own alternative resource mortality function. If
your function is called "myResourceMort" then you register it in a MizerParams
object params with
params <- setRateFunction(params, "ResourceMort", "myResourceMort")
Your function will then be called instead of mizerResourceMort(), with the
same arguments.
See Also
Other rate functions:
getDiffusion(),
getEGrowth(),
getERepro(),
getEReproAndGrowth(),
getEncounter(),
getFMort(),
getFMortGear(),
getFeedingLevel(),
getFlux(),
getFluxGradient(),
getMort(),
getPredMort(),
getPredRate(),
getRDD(),
getRDI(),
getRates()
Examples
params <- NS_params
# With constant fishing effort for all gears for 20 time steps
sim <- project(params, t_max = 20, effort = 0.5)
# Get resource mortality at one time step
getResourceMort(params, n = N(sim)[15, , ], n_pp = NResource(sim)[15, ])
Calculate the mean maximum weight of the community
Description
Calculates the mean maximum weight of the community. This can be calculated
by numbers or biomass. The calculation is the sum of the w_inf * abundance
of each species, divided by the total abundance community, where abundance is
either in biomass or numbers. You can specify minimum and maximum weight or
length range for the species. Lengths take precedence over weights (i.e. if
both min_l and min_w are supplied, only min_l will be used). You can also
specify the species to be used in the calculation.
Usage
getMeanMaxWeight(object, species = NULL, measure = "both", ...)
Arguments
object |
A MizerSim or MizerParams object |
species |
The species to be selected. Optional. By default all target species are selected. A vector of species names, or a numeric vector with the species indices, or a logical vector indicating for each species whether it is to be selected (TRUE) or not. |
measure |
The measure to return. Can be 'numbers', 'biomass' or 'both' |
... |
Arguments passed on to
|
Value
Depends on the measure argument. If measure = “both”
then you get a matrix with two columns, one with values by numbers,
the other with values by biomass at each saved time step (or a named
vector with two entries for MizerParams). If measure =
“numbers” or “biomass” you get a vector of the respective values
at each saved time step (or a single value for MizerParams).
See Also
Other functions for calculating indicators:
getCommunitySlope(),
getMeanWeight(),
getProportionOfLargeFish()
Examples
mmw <- getMeanMaxWeight(NS_sim)
years <- c("1967", "2010")
mmw[years, ]
getMeanMaxWeight(NS_sim, species=c("Herring","Sprat","N.pout"))[years, ]
getMeanMaxWeight(NS_sim, min_w = 10, max_w = 5000)[years, ]
getMeanMaxWeight(NS_params)
Calculate the mean weight of the community
Description
Calculates the mean weight of the community. This is simply the total biomass of the community divided by the abundance in numbers. You can specify minimum and maximum weight or length range for the species. Lengths take precedence over weights (i.e. if both min_l and min_w are supplied, only min_l will be used). You can also specify the species to be used in the calculation.
Usage
getMeanWeight(object, species = NULL, ...)
Arguments
object |
A MizerSim or MizerParams object |
species |
The species to be selected. Optional. By default all target species are selected. A vector of species names, or a numeric vector with the species indices, or a logical vector indicating for each species whether it is to be selected (TRUE) or not. |
... |
Arguments passed on to
|
Value
A vector containing the mean weight of the community through time,
or a single value if called with a MizerParams object.
See Also
Other functions for calculating indicators:
getCommunitySlope(),
getMeanMaxWeight(),
getProportionOfLargeFish()
Examples
mean_weight <- getMeanWeight(NS_sim)
years <- c("1967", "2010")
mean_weight[years]
getMeanWeight(NS_sim, species = c("Herring", "Sprat", "N.pout"))[years]
getMeanWeight(NS_sim, min_w = 10, max_w = 5000)[years]
getMeanWeight(NS_params)
Get total mortality rate
Description
Calculates the total mortality rate \mu_i(w) (in units 1/year) on each
species by size from predation mortality, background mortality and fishing
mortality for a single time step.
Usage
getMort(object, ...)
Arguments
object |
A MizerParams or MizerSim object. |
... |
Additional arguments that depend on the class of For a MizerParams object:
For a MizerSim object:
|
Details
If your model contains additional components that you added with
setComponent() and for which you specified a mort_fun function then
the mortality inflicted by these components will be included in the returned
value.
Value
-
MizerParams: AnArraySpeciesBySizeobject (species x size) with the total mortality rates. -
MizerSim: AnArrayTimeBySpeciesBySizeobject (time step x species x size) with the total mortality rates at every time step. Ifdrop = TRUEthen dimensions of length 1 will be removed.
Your own mortality function
By default getMort() calls mizerMort(). However you can
replace this with your own alternative mortality function. If
your function is called "myMort" then you register it in a MizerParams
object params with
params <- setRateFunction(params, "Mort", "myMort")
Your function will then be called instead of mizerMort(), with the
same arguments.
See Also
Other rate functions:
getDiffusion(),
getEGrowth(),
getERepro(),
getEReproAndGrowth(),
getEncounter(),
getFMort(),
getFMortGear(),
getFeedingLevel(),
getFlux(),
getFluxGradient(),
getPredMort(),
getPredRate(),
getRDD(),
getRDI(),
getRates(),
getResourceMort()
Examples
params <- NS_params
# Project with constant fishing effort for all gears for 20 time steps
sim <- project(params, t_max = 20, effort = 0.5)
# Get the total mortality at a particular time step
mort <- getMort(params, n = N(sim)[15, , ], n_pp = NResource(sim)[15, ],
t = 15, effort = 0.5)
# Mortality rate at this time for Sprat of size 2g
mort["Sprat", "2"]
Calculate the number of individuals within a size range
Description
Calculates the number of individuals within user-defined size limits. The default option is to use the whole size range. You can specify minimum and maximum weight or lengths for the species. Lengths take precedence over weights (i.e. if both min_l and min_w are supplied, only min_l will be used)
Usage
getN(object, ...)
Arguments
object |
An object of class |
... |
Arguments passed on to
|
Value
If called with a MizerParams object, a named vector with the numbers
for each species in the model. If called with a MizerSim object, a
ArrayTimeBySpecies object (time x species) containing the numbers at
each time step for all species.
See Also
Other summary functions:
getBiomass(),
getDiet(),
getGrowthCurves(),
getSSB(),
getTrophicLevel(),
getTrophicLevelBySpecies(),
getYield(),
getYieldGear()
Examples
numbers <- getN(NS_sim)
numbers["1972", "Herring"]
# The above gave a huge number, because that included all the larvae.
# The number of Herrings between 10g and 1kg is much smaller.
numbers <- getN(NS_sim, min_w = 10, max_w = 1000)
numbers["1972", "Herring"]
Extract the model state from a simulation
Description
A MizerParams object describes the state of the ecosystem: its species
parameters, size grid, rate functions, and the current abundances stored in
the initial_n, initial_n_pp, initial_n_other, and initial_effort
slots. getParams() extracts that state from a MizerSim object, averaged
over a chosen time range (or at a single time point).
Usage
getParams(sim, time_range, geometric_mean = FALSE)
Arguments
sim |
A |
time_range |
The time range to average the abundances over. Can be a vector of values, a vector of min and max time, or a single value. Only the range of times is relevant, i.e., all times between the smallest and largest will be selected. Default is the final time step. |
geometric_mean |
|
Details
When no time_range is given, the state at the final time step is returned.
Use initialParams() or finalParams() as convenient shorthand for the
state at the initial and final time respectively.
The abundances set in the returned MizerParams object are averages over the
selected time range. By default this is an arithmetic mean; set
geometric_mean = TRUE to use a geometric mean instead (this does not affect
the effort or other components, which are always averaged arithmetically).
Value
A MizerParams object with initial_n, initial_n_pp,
initial_n_other, and initial_effort set to the (averaged) values from
the simulation.
See Also
initialParams(), finalParams()
Examples
sim <- project(NS_params, t_max = 20, effort = 0.5)
# Extract state at a specific time
params_2010 <- getParams(sim, time_range = 10)
# Extract state averaged over the last 10 years
params_avg <- getParams(sim, time_range = c(10, 20))
Get available energy
Description
This is deprecated and is no longer used by the mizer project() method.
Calculates the amount E_{a,i}(w) of food exposed to each predator as
a function of predator size.
Usage
getPhiPrey(object, n, n_pp, ...)
Arguments
object |
An MizerParams object |
n |
A matrix of species abundances (species x size) |
n_pp |
A vector of the background abundance by size |
... |
Other arguments (currently unused) |
Value
A two dimensional array (predator species x predator size)
equal to getEncounter(object, n, n_pp) / getSearchVolume(object).
See Also
Get total predation mortality rate
Description
Calculates the total predation mortality rate \mu_{p,i}(w_p) (in units
of 1/year) on each prey species by prey size:
\mu_{p.i}(w_p) = \sum_j {\tt pred\_rate}_j(w_p)\, \theta_{ji}.
The predation rate pred_rate is returned by getPredRate().
Usage
getPredMort(object, ...)
Arguments
object |
A MizerParams or MizerSim object. |
... |
Additional arguments that depend on the class of For a MizerParams object:
For a MizerSim object:
|
Value
-
MizerParams: AnArraySpeciesBySizeobject (prey species x prey size) with the predation mortality rates. -
MizerSim: AnArrayTimeBySpeciesBySizeobject (time step x prey species x prey size) with the predation mortality at every time step. Ifdrop = TRUEthen dimensions of length 1 will be removed.
Your own predation mortality function
By default getPredMort() calls mizerPredMort(). However you can
replace this with your own alternative predation mortality function. If
your function is called "myPredMort" then you register it in a MizerParams
object params with
params <- setRateFunction(params, "PredMort", "myPredMort")
Your function will then be called instead of mizerPredMort(), with the
same arguments.
See Also
Other rate functions:
getDiffusion(),
getEGrowth(),
getERepro(),
getEReproAndGrowth(),
getEncounter(),
getFMort(),
getFMortGear(),
getFeedingLevel(),
getFlux(),
getFluxGradient(),
getMort(),
getPredRate(),
getRDD(),
getRDI(),
getRates(),
getResourceMort()
Examples
params <- NS_params
# Predation mortality in initial state
M2 <- getPredMort(params)
str(M2)
# With constant fishing effort for all gears for 20 time steps
sim <- project(params, t_max = 20, effort = 0.5)
# Get predation mortality at one time step
M2 <- getPredMort(params, n = N(sim)[15, , ], n_pp = NResource(sim)[15, ])
# Get predation mortality at all saved time steps
M2 <- getPredMort(sim)
str(M2)
# Get predation mortality over the years 15 - 20
M2 <- getPredMort(sim, time_range = c(15, 20))
Get predation rate
Description
Calculates the potential rate (in units 1/year) at which a prey individual of
a given size w is killed by predators from species j. In formulas
{\tt pred\_rate}_j(w_p) = \int \phi_j(w,w_p) (1-f_j(w))
\gamma_j(w) N_j(w) \, dw.
This potential rate is used in getPredMort() to
calculate the realised predation mortality rate on the prey individual.
Usage
getPredRate(object, ...)
Arguments
object |
A MizerParams or MizerSim object. |
... |
Additional arguments that depend on the class of For a MizerParams object:
For a MizerSim object:
|
Value
-
MizerParams: AnArraySpeciesBySizeobject (predator species x prey size), where the prey size runs over fish community plus resource spectrum. -
MizerSim: AnArrayTimeBySpeciesBySizeobject (time step x predator species x prey size) with the predation rates at every time step. Ifdrop = TRUEthen dimensions of length 1 will be removed.
Your own predation rate function
By default getPredRate() calls mizerPredRate(). However you can
replace this with your own alternative predation rate function. If
your function is called "myPredRate" then you register it in a MizerParams
object params with
params <- setRateFunction(params, "PredRate", "myPredRate")
Your function will then be called instead of mizerPredRate(), with
the same arguments.
See Also
Other rate functions:
getDiffusion(),
getEGrowth(),
getERepro(),
getEReproAndGrowth(),
getEncounter(),
getFMort(),
getFMortGear(),
getFeedingLevel(),
getFlux(),
getFluxGradient(),
getMort(),
getPredMort(),
getRDD(),
getRDI(),
getRates(),
getResourceMort()
Examples
params <- NS_params
# Predation rate in initial state
pred_rate <- getPredRate(params)
str(pred_rate)
# With constant fishing effort for all gears for 20 time steps
sim <- project(params, t_max = 20, effort = 0.5)
# Get the feeding level at one time step
pred_rate <- getPredRate(params, n = N(sim)[15, , ],
n_pp = NResource(sim)[15, ], t = 15)
Calculate the proportion of large fish
Description
Calculates the proportion of large fish in a MizerSim or MizerParams
object within user defined size limits. The default option is to use the
whole size range. You can specify minimum and maximum size ranges for the
species and also the threshold size for large fish. Sizes can be expressed
as weight or length. Lengths take precedence over weights (i.e. if both
min_l and min_w are supplied, only min_l will be used, and if
threshold_l is supplied it takes precedence over threshold_w). You can
also specify the species to be used in the calculation. This function can be
used to calculate the Large Fish Index. The proportion is based on either
abundance or biomass.
Usage
getProportionOfLargeFish(
object,
species = NULL,
threshold_w = 100,
threshold_l = NULL,
biomass_proportion = TRUE,
...
)
Arguments
object |
A MizerSim or MizerParams object |
species |
The species to be selected. Optional. By default all target species are selected. A vector of species names, or a numeric vector with the species indices, or a logical vector indicating for each species whether it is to be selected (TRUE) or not. |
threshold_w |
The weight used as the cutoff between large and small fish. Default value is 100. |
threshold_l |
The length used as the cutoff between large and small
fish. If supplied, this takes precedence over |
biomass_proportion |
A boolean value. If TRUE the proportion calculated is based on biomass, if FALSE it is based on numbers of individuals. Default is TRUE. |
... |
Arguments passed on to
|
Value
A vector containing the proportion of large fish through time, or a
single value if called with a MizerParams object.
See Also
Other functions for calculating indicators:
getCommunitySlope(),
getMeanMaxWeight(),
getMeanWeight()
Examples
lfi <- getProportionOfLargeFish(NS_sim, min_w = 10, max_w = 5000,
threshold_w = 500)
years <- c("1972", "2010")
lfi[years]
getProportionOfLargeFish(NS_sim)[years]
getProportionOfLargeFish(NS_sim, species=c("Herring","Sprat","N.pout"))[years]
getProportionOfLargeFish(NS_sim, min_w = 10, max_w = 5000)[years]
getProportionOfLargeFish(NS_sim, min_w = 10, max_w = 5000,
threshold_w = 500, biomass_proportion = FALSE)[years]
getProportionOfLargeFish(NS_params)
Get density dependent reproduction rate
Description
Calculates the density dependent rate of egg production R_i (units
1/year) for each species. This is the flux entering the smallest size class
of each species. The density dependent rate is the density independent
rate obtained with getRDI() after it has been put through the
density dependence function. This is the Beverton-Holt function
BevertonHoltRDD() by default, but this can be changed. See
setReproduction() for more details.
Usage
getRDD(object, ...)
Arguments
object |
A MizerParams or MizerSim object. |
... |
Additional arguments that depend on the class of For a MizerParams object:
For a MizerSim object:
|
Value
-
MizerParams: A numeric vector the length of the number of species. -
MizerSim: AnArrayTimeBySpeciesobject (time x species).
See Also
Other rate functions:
getDiffusion(),
getEGrowth(),
getERepro(),
getEReproAndGrowth(),
getEncounter(),
getFMort(),
getFMortGear(),
getFeedingLevel(),
getFlux(),
getFluxGradient(),
getMort(),
getPredMort(),
getPredRate(),
getRDI(),
getRates(),
getResourceMort()
Examples
params <- NS_params
# Project with constant fishing effort for all gears for 20 time steps
sim <- project(params, t_max = 20, effort = 0.5)
# Get the rate at a particular time step
getRDD(params, n = N(sim)[15, , ], n_pp = NResource(sim)[15, ], t = 15)
Get density independent rate of egg production
Description
Calculates the density-independent rate of total egg production
R_{di} (units 1/year) before density dependence, by species.
Usage
getRDI(object, ...)
Arguments
object |
A MizerParams or MizerSim object. |
... |
Additional arguments that depend on the class of For a MizerParams object:
For a MizerSim object:
|
Details
This rate is obtained by taking the per capita rate E_r(w)\psi(w) at
which energy is invested in reproduction, as calculated by getERepro(),
multiplying it by the number of individualsN(w) and integrating over
all sizes w and then multiplying by the reproductive efficiency
\epsilon and dividing by the egg size w_min, and by a factor of two
to account for the two sexes:
R_{di} = \frac{\epsilon}{2 w_{min}} \int N(w) E_r(w) \psi(w) \, dw
Used by getRDD() to calculate the actual, density dependent rate.
See setReproduction() for more details.
Value
-
MizerParams: A numeric vector the length of the number of species. -
MizerSim: AnArrayTimeBySpeciesobject (time x species).
Your own reproduction function
By default getRDI() calls mizerRDI(). However you can
replace this with your own alternative reproduction function. If
your function is called "myRDI" then you register it in a MizerParams
object params with
params <- setRateFunction(params, "RDI", "myRDI")
Your function will then be called instead of mizerRDI(), with the
same arguments. For an example of an alternative reproduction function
see constantEggRDI().
See Also
Other rate functions:
getDiffusion(),
getEGrowth(),
getERepro(),
getEReproAndGrowth(),
getEncounter(),
getFMort(),
getFMortGear(),
getFeedingLevel(),
getFlux(),
getFluxGradient(),
getMort(),
getPredMort(),
getPredRate(),
getRDD(),
getRates(),
getResourceMort()
Examples
params <- NS_params
# Project with constant fishing effort for all gears for 20 time steps
sim <- project(params, t_max = 20, effort = 0.5)
# Get the density-independent reproduction rate at a particular time step
getRDI(params, n = N(sim)[15, , ], n_pp = NResource(sim)[15, ], t = 15)
Get all rates
Description
Calls other rate functions in sequence and collects the results in a list. The rates returned are encounter, feeding level, energy for growth and reproduction, predation rate, predation mortality, and resource mortality. The purpose of this function is to provide a convenient way to get all the rates at once, and to ensure that they are all calculated at the same time step with the same inputs. The rates are returned in a list with the same names as the rate functions that calculate them, so for example the encounter rate is returned in the list element named "encounter" and is calculated with the getEncounter() function.
Usage
getRates(
params,
n = initialN(params),
n_pp = initialNResource(params),
n_other = initialNOther(params),
effort,
t = 0,
...
)
Arguments
params |
A MizerParams object |
n |
A matrix of species abundances (species x size). |
n_pp |
A vector of the resource abundance by size |
n_other |
A list of abundances for other dynamical components of the ecosystem |
effort |
The effort for each fishing gear |
t |
The time for which to do the calculation (Not used by standard mizer rate functions but useful for extensions with time-dependent parameters.) |
... |
Unused |
Details
When mizer needs to calculate the rates during a simulation it does not use
this function but instead the faster projectRates().
See Also
Other rate functions:
getDiffusion(),
getEGrowth(),
getERepro(),
getEReproAndGrowth(),
getEncounter(),
getFMort(),
getFMortGear(),
getFeedingLevel(),
getFlux(),
getFluxGradient(),
getMort(),
getPredMort(),
getPredRate(),
getRDD(),
getRDI(),
getResourceMort()
Examples
rates <- getRates(NS_params)
names(rates)
identical(rates$encounter, getEncounter(NS_params))
Get the registered mizer extension chain
Description
Get the registered mizer extension chain
Usage
getRegisteredExtensions()
Value
A named character vector giving the maximal extension chain registered for this R session.
See Also
"Using mizer extension packages":
vignette("using-extension-packages", package = "mizer")
Other extension tools:
NOther(),
clearExtensionChain(),
coerceToExtensionClass(),
initialNOther<-(),
recordExtension(),
registerExtension(),
registerExtensions(),
setComponent(),
setRateFunction()
Get reproduction level
Description
The reproduction level is the ratio between the density-dependent reproduction rate and the maximal reproduction rate.
Usage
getReproductionLevel(params)
Arguments
params |
A MizerParams object |
Value
A named vector with the reproduction level for each species.
Examples
getReproductionLevel(NS_params)
# The reproduction level can be changed without changing the steady state:
params <- setBevertonHolt(NS_params, reproduction_level = 0.9)
getReproductionLevel(params)
# The result is the ratio of RDD and R_max
identical(getRDD(params) / species_params(params)$R_max,
getReproductionLevel(params))
Determine reproduction rate needed for initial egg abundance
Description
Determine reproduction rate needed for initial egg abundance
Usage
getRequiredRDD(params, ...)
Arguments
params |
A MizerParams object |
... |
Unused. |
Value
A vector of reproduction rates for all species
Deprecated functions for getting resource parameters
Description
Use resource_dynamics(),
resource_level(), resource_rate() and resource_capacity() instead.
Usage
getResourceDynamics(params)
getResourceLevel(params)
getResourceRate(params)
getResourceCapacity(params)
Arguments
params |
A MizerParams object |
Value
Name of the function that determines the resource dynamics
Get predation mortality rate for resource
Description
Calculates the predation mortality rate \mu_p(w) on the resource
spectrum by resource size (in units 1/year).
Usage
getResourceMort(
params,
n = initialN(params),
n_pp = initialNResource(params),
n_other = initialNOther(params),
t = 0,
...
)
Arguments
params |
A MizerParams object |
n |
A matrix of species abundances (species x size). |
n_pp |
A vector of the resource abundance by size |
n_other |
A list of abundances for other dynamical components of the ecosystem |
t |
The time for which to do the calculation (Not used by standard mizer rate functions but useful for extensions with time-dependent parameters.) |
... |
Unused |
Value
A vector of mortality rate by resource size.
Your own resource mortality function
By default getResourceMort() calls mizerResourceMort(). However you can
replace this with your own alternative resource mortality function. If
your function is called "myResourceMort" then you register it in a MizerParams
object params with
params <- setRateFunction(params, "ResourceMort", "myResourceMort")
Your function will then be called instead of mizerResourceMort(), with the
same arguments.
See Also
Other rate functions:
getDiffusion(),
getEGrowth(),
getERepro(),
getEReproAndGrowth(),
getEncounter(),
getFMort(),
getFMortGear(),
getFeedingLevel(),
getFlux(),
getFluxGradient(),
getMort(),
getPredMort(),
getPredRate(),
getRDD(),
getRDI(),
getRates()
Examples
params <- NS_params
# With constant fishing effort for all gears for 20 time steps
sim <- project(params, t_max = 20, effort = 0.5)
# Get resource mortality at one time step
getResourceMort(params, n = N(sim)[15, , ], n_pp = NResource(sim)[15, ])
Calculate the SSB of species
Description
Calculates the spawning stock biomass (SSB) for each species. For a
MizerSim object this is returned for every saved time; for a
MizerParams object it is calculated from the initial state. SSB is the
total mass of all mature individuals.
Usage
getSSB(object)
Arguments
object |
An object of class |
Value
If called with a MizerParams object, a named vector with the SSB in
grams for each species in the model. If called with a MizerSim object, a
ArrayTimeBySpecies object (time x species) containing the SSB in grams
at each time step for all species.
See Also
Other summary functions:
getBiomass(),
getDiet(),
getGrowthCurves(),
getN(),
getTrophicLevel(),
getTrophicLevelBySpecies(),
getYield(),
getYieldGear()
Examples
ssb <- getSSB(NS_sim)
ssb[c("1972", "2010"), c("Herring", "Cod")]
Extract the projection parameters used to produce a simulation
Description
Returns the named list of arguments passed to project() or
projectToSteady() when producing this MizerSim object, such as
method and dt. Returns an empty list for simulations produced by
older versions of mizer.
Usage
getSimParams(sim)
Arguments
sim |
A MizerSim object |
Value
A named list of projection parameters.
Examples
sim <- project(NS_params, t_max = 0.1, dt = 0.05, method = "predictor-corrector")
getSimParams(sim)
Times for which simulation results are available
Description
Times for which simulation results are available
Usage
getTimes(sim)
Arguments
sim |
A MizerSim object |
Value
A numeric vector of the times (in years) at which simulation results have been stored in the MizerSim object.
Examples
getTimes(NS_sim)
Get trophic level of individuals at size
Description
Calculates the trophic level of individuals of each species at each size,
assuming the system is in a steady state. The trophic level of an individual
is defined as 1 more than the consumption-rate-weighted average trophic level
of all the prey it has consumed during its lifetime up to the current size.
The resource is given a size-dependent trophic level (see below).
Usage
getTrophicLevel(
params,
n = initialN(params),
n_pp = initialNResource(params),
n_other = initialNOther(params),
w_R = 1e-10,
beta_R = 1000,
...
)
Arguments
params |
A MizerParams object. |
n |
A matrix of species abundances (species x size). Defaults to
the initial abundances stored in |
n_pp |
A vector of the resource abundance by size. Defaults to the
initial resource abundance stored in |
n_other |
A named list of the abundances of other dynamical
components. Defaults to the initial values stored in |
w_R |
An average size (in grams) of primary producers in the resource
spectrum, used to set the size-dependent resource trophic level. Defaults
to |
beta_R |
An average predator/prey mass ratio for the resource spectrum,
used to set the size-dependent resource trophic level. Must be greater than
|
... |
Unused |
Details
In the traditional non-size-resolved approach, all individuals of a species
have the same diet composition D_{ij}, defined as the proportion of
total biomass intake of species i that comes from species j.
The trophic levels then satisfy
T_i = 1 + \sum_j D_{ij}\,T_j,
which is solved as a linear system (I - D)\,\mathbf{T} = \mathbf{1}.
In mizer, diet composition changes as an individual grows, so we must
integrate over the individual's lifetime. Assuming a steady state so that
the growth rate g_i(w) and prey densities depend only on size and not
on time, we can replace the integral over time since birth by an integral
over weight using dt = dw / g_i(w). The trophic level
T_i(w) of an individual of species i at weight w is
then
T_i(w) = 1 + \frac{
\int_{w_0}^{w} \frac{1}{g_i(w')} \sum_j \int r_{ij}(w', w_p)\, T_j(w_p)\, dw_p\, dw'
}{
\int_{w_0}^{w} \frac{1}{g_i(w')} \sum_j \int r_{ij}(w', w_p)\, dw_p\, dw'
},
where w_0 is the egg size and r_{ij}(w, w_p) is the rate at
which a predator of species i at weight w consumes biomass from
prey species j at weight w_p:
r_{ij}(w, w_p) = \theta_{ij}\,\gamma_i(w)\,(1 - f_i(w))\,\phi_i(w/w_p)\,
N_j(w_p)\,w_p.
The sum over j runs over all species and the resource. The resource is
assigned a size-dependent trophic level
T_R(w) = \max\left(1,\; 1 + \frac{\log(w / w_R)}{\log(\beta_R)}\right),
where w_R is an average size of primary producers (which therefore have
trophic level 1) and \beta_R is an average predator/prey mass ratio for
the resource (for example zooplankton). This adds one trophic level for each
factor of \beta_R increase in resource size, with a floor at 1 so that
the resource trophic level never drops below the primary-producer level.
Both the numerator and the denominator (which equals the total biomass
consumed over the predator's lifetime from egg size to current weight
w) therefore include the resource.
This equation can be viewed as a linear system
(I - D)\,\mathbf{T} = \mathbf{1} in which the entries of
\mathbf{T} are indexed by (i, w) and the matrix D encodes
the lifetime-integrated diet composition. The system is solved iteratively
from small to large sizes, exploiting the fact that prey are typically much
smaller than the predator (large predator-to-prey mass ratio), so that the
trophic levels of all relevant prey sizes are already known when computing
T_i(w).
Value
An ArraySpeciesBySize object (species x size) with the trophic
level of individuals at each size. Entries below the egg size of each
species are NA.
See Also
Other summary functions:
getBiomass(),
getDiet(),
getGrowthCurves(),
getN(),
getSSB(),
getTrophicLevelBySpecies(),
getYield(),
getYieldGear()
Examples
tl <- getTrophicLevel(NS_params)
plot(tl)
Get mean trophic level of each species
Description
Calculates the consumption-rate-weighted mean trophic level of each species,
defined as
T_i = \frac{\int r_i(w)\,N_i(w)\,T_i(w)\,dw}
{\int r_i(w)\,N_i(w)\,dw},
where r_i(w) = (1 - f_i(w))\,E_i(w) is the consumption rate of an
individual of species i at weight w, N_i(w) is the
abundance density, and T_i(w) is the size-resolved trophic level
from getTrophicLevel(). As in getTrophicLevel(), the resource is given a
size-dependent trophic level controlled by the w_R and beta_R arguments.
Usage
getTrophicLevelBySpecies(
params,
n = initialN(params),
n_pp = initialNResource(params),
n_other = initialNOther(params),
w_R = 1e-10,
beta_R = 1000,
...
)
Arguments
params |
A MizerParams object. |
n |
A matrix of species abundances (species x size). Defaults to
the initial abundances stored in |
n_pp |
A vector of the resource abundance by size. Defaults to the
initial resource abundance stored in |
n_other |
A named list of the abundances of other dynamical
components. Defaults to the initial values stored in |
w_R |
An average size (in grams) of primary producers in the resource
spectrum, used to set the size-dependent resource trophic level. Defaults
to |
beta_R |
An average predator/prey mass ratio for the resource spectrum,
used to set the size-dependent resource trophic level. Must be greater than
|
... |
Unused |
Value
A named vector with the mean trophic level for each species.
See Also
Other summary functions:
getBiomass(),
getDiet(),
getGrowthCurves(),
getN(),
getSSB(),
getTrophicLevel(),
getYield(),
getYieldGear()
Examples
getTrophicLevelBySpecies(NS_params)
Calculate the rate at which biomass of each species is fished
Description
This yield rate is given in grams per year. It is calculated at each time step saved in the MizerSim object.
Usage
getYield(object)
Arguments
object |
An object of class |
Details
The yield rate y_i(t) for species i at time t is defined as
y_i(t)=\int\mu_{f.i}(w, t)N_i(w, t)w dw
where \mu_{f.i}(w, t) is the fishing mortality of an individual of
species i and weight w at time t and N_i(w, t) is the
abundance density of such individuals. The factor of w converts the
abundance density into a biomass density and the integral aggregates the
contribution from all sizes.
The total catch in a time period from t_1 to t_2 is the integral
of the yield rate over that period:
C = \int_{t_1}^{t2}y_i(t)dt
In practice, as the yield rate is only available
at the saved times, one can only approximate this integral by averaging over
the available yield rates during the time period and multiplying by the time
period. The less the yield changes between the saved values, the more
accurate this approximation is. So the approximation can be improved by
saving simulation results at smaller intervals, using the t_save argument
to project(). But this is only a concern if abundances change quickly
during the time period of interest.
Value
If called with a MizerParams object, a named numeric vector with
the yield rate in grams per year for each species in the model. If called
with a MizerSim object, an ArrayTimeBySpecies object (time x species)
containing the yield rate in grams per year at each saved time step.
See Also
Other summary functions:
getBiomass(),
getDiet(),
getGrowthCurves(),
getN(),
getSSB(),
getTrophicLevel(),
getTrophicLevelBySpecies(),
getYieldGear()
Examples
yield <- getYield(NS_sim)
yield[c("1972", "2010"), c("Herring", "Cod")]
# Running simulation for another year, saving intermediate time steps
params <- finalParams(NS_sim)
sim <- project(params, t_save = 0.1, t_max = 1,
t_start = 2010, progress_bar = FALSE)
# The yield rate for Herring decreases during the year
getYield(sim)[, "Herring"]
# We approximate the total catch in the year by averaging over the year
sum(getYield(sim)[1:10, "Herring"] / 10)
Calculate the rate at which biomass of each species is fished by each gear
Description
This yield rate is given in grams per year. It is calculated at each time step saved in the MizerSim object.
Usage
getYieldGear(object)
Arguments
object |
An object of class |
Details
For details of how the yield rate is defined see the help page of
getYield().
Value
If called with a MizerParams object, an array (gear x species) with the yield rate in grams per year from each gear for each species in the model. If called with a MizerSim object, an array (time x gear x species) containing the yield rate at each time step.
See Also
Other summary functions:
getBiomass(),
getDiet(),
getGrowthCurves(),
getN(),
getSSB(),
getTrophicLevel(),
getTrophicLevelBySpecies(),
getYield()
Examples
yield <- getYieldGear(NS_sim)
dim(yield)
yield["1972", , "Herring"]
Alias for getMort()
Description
An alias provided for backward compatibility with mizer version <= 1.0
Usage
getZ(object, ...)
Arguments
object |
A MizerParams or MizerSim object. |
... |
Additional arguments that depend on the class of For a MizerParams object:
For a MizerSim object:
|
Details
If your model contains additional components that you added with
setComponent() and for which you specified a mort_fun function then
the mortality inflicted by these components will be included in the returned
value.
Value
-
MizerParams: AnArraySpeciesBySizeobject (species x size) with the total mortality rates. -
MizerSim: AnArrayTimeBySpeciesBySizeobject (time step x species x size) with the total mortality rates at every time step. Ifdrop = TRUEthen dimensions of length 1 will be removed.
Your own mortality function
By default getMort() calls mizerMort(). However you can
replace this with your own alternative mortality function. If
your function is called "myMort" then you register it in a MizerParams
object params with
params <- setRateFunction(params, "Mort", "myMort")
Your function will then be called instead of mizerMort(), with the
same arguments.
See Also
Other rate functions:
getDiffusion(),
getEGrowth(),
getERepro(),
getEReproAndGrowth(),
getEncounter(),
getFMort(),
getFMortGear(),
getFeedingLevel(),
getFlux(),
getFluxGradient(),
getPredMort(),
getPredRate(),
getRDD(),
getRDI(),
getRates(),
getResourceMort()
Examples
params <- NS_params
# Project with constant fishing effort for all gears for 20 time steps
sim <- project(params, t_max = 20, effort = 0.5)
# Get the total mortality at a particular time step
mort <- getMort(params, n = N(sim)[15, , ], n_pp = NResource(sim)[15, ],
t = 15, effort = 0.5)
# Mortality rate at this time for Sprat of size 2g
mort["Sprat", "2"]
Get the size grid for an ArrayResourceBySize object
Description
Internal helper that returns the full prey/resource size grid
params@w_full, or the numeric vector parsed from the names of x if no
params is attached.
Usage
get_ArrayResourceBySize_w(x)
Arguments
x |
An |
Value
A numeric vector giving the size represented by each element.
Get the size grid for an ArraySpeciesBySize object
Description
Internal helper that returns the consumer size grid params@w or the full
prey/resource size grid params@w_full, depending on the number of columns
in the array.
Usage
get_ArraySpeciesBySize_w(x)
Arguments
x |
An |
Value
A numeric vector giving the size represented by each column. When the
array is tagged as a bin average (representation = "average") and the
model uses second-order bin-averaging (second_order_w[["bin_average"]]),
the geometric bin centres are returned instead of the left bin edges, so
that bin-averaged quantities are drawn at the size where they actually live
(see bin_midpoints()). Point-valued quantities and first-order models are
unaffected, keeping default plots unchanged.
Get the size grid for an ArrayTimeBySpeciesBySize object
Description
Internal helper, the three-dimensional analogue of
get_ArraySpeciesBySize_w(). Returns the geometric bin centres (see
bin_midpoints()) when the array is tagged as a bin average and the model
uses second-order bin-averaging, otherwise the grid nodes read from the size
dimension names. Falls back to the dimension names when no params is
attached.
Usage
get_ArrayTimeBySpeciesBySize_w(x)
Arguments
x |
An |
Value
A numeric vector giving the size represented by each size slice.
Get default value for f0
Description
Fills in any missing values for f0 so that if the prey abundance was
described by the power law \kappa w^{-\lambda} then the encounter rate
coming from the given gamma parameter would lead to the feeding level
f_0. This is thus doing the inverse of get_gamma_default().
Only for internal use.
Usage
get_f0_default(params)
Arguments
params |
A MizerParams object |
Details
For species for which no value for gamma is specified in the species
parameter data frame, the f0 values is kept as provided in the species
parameter data frame or it is set to 0.6 if it is not provided.
See the Target Feeding Level section of the "Calculation of Default Parameter Values" vignette for the mathematical derivation.
Value
A vector with the values of f0 for all species
See Also
Other functions calculating defaults:
get_gamma_default(),
get_h_default(),
get_ks_default()
Get default value for gamma
Description
Fills in any missing values for gamma so that fish feeding on a resource
spectrum described by the power law \kappa w^{-\lambda} achieve a
feeding level f_0. Only for internal use.
Usage
get_gamma_default(params)
Arguments
params |
A MizerParams object |
Details
See the Search Volume Coefficient section of the "Calculation of Default Parameter Values" vignette for the mathematical derivation.
Value
A vector with the values of gamma for all species
See Also
Other functions calculating defaults:
get_f0_default(),
get_h_default(),
get_ks_default()
Get default value for h
Description
Sets h so that the species reaches maturity size w_mat at the maturity
age age_mat if it feeds at feeding level f0.
Usage
get_h_default(params)
Arguments
params |
A MizerParams object or a species parameter data frame |
Details
If age_mat is missing in the species parameter data frame, then it is
calculated from the von Bertalanffy growth curve parameters k_vb and
(optionally t0) taken from the species parameter data frame. This is not
reliable and a warning is issued.
If no growth information is given at all for a species, the default is set
to h = 30.
See the Maximum Intake Rate Coefficient section of the "Calculation of Default Parameter Values" vignette for the mathematical derivation.
Value
A vector with the values of h for all species
See Also
Other functions calculating defaults:
get_f0_default(),
get_gamma_default(),
get_ks_default()
Calculate initial population abundances
Description
This function uses the model parameters and other parameters to calculate initial values for the species number densities. These initial abundances are currently quite arbitrary and not close to the steady state. We intend to improve this in the future.
Usage
get_initial_n(params, n0_mult = NULL, a = 0.35)
Arguments
params |
The model parameters. An object of type MizerParams. |
n0_mult |
Multiplier for the abundance at size 0 when using defaults
edition 1. If not supplied, |
a |
A parameter with a default value of 0.35. |
Value
An ArraySpeciesBySize object (species x size) of population abundances.
Examples
init_n <- get_initial_n(NS_params)
Get default value for ks
Description
Fills in any missing values for ks so that the critical feeding level needed
to sustain the species is as specified in the fc column in the species
parameter data frame. If that column is not provided the default critical
feeding level f_c = 0.2 is used.
Usage
get_ks_default(params)
Arguments
params |
A MizerParams object |
Details
See the Standard Metabolic Rate Coefficient section of the "Calculation of Default Parameter Values" vignette for the mathematical derivation.
Value
A vector with the values of ks for all species
See Also
Other functions calculating defaults:
get_f0_default(),
get_gamma_default(),
get_h_default()
Get values from feeding kernel function
Description
This involves finding the feeding kernel function for each species, using the pred_kernel_type parameter in the species_params data frame, checking that it is valid and all its arguments are contained in the species_params data frame, and then calling this function with the ppmr vector.
Usage
get_phi(species_params, ppmr)
Arguments
species_params |
A species parameter data frame |
ppmr |
Values of the predator/prey mass ratio at which to evaluate the predation kernel function |
Value
An array (species x ppmr) with the values of the predation kernel function
Extract one saved simulation state for a rate calculation
Description
Internal helper used by MizerSim rate methods to rebuild the single-time
inputs expected by MizerParams rate methods.
Usage
get_sim_rate_slice(sim, time_idx)
Arguments
sim |
A |
time_idx |
Integer index of the saved time step to extract. |
Value
A list with entries n, n_pp, n_other, effort, and t.
Get selected saved time steps for a simulation rate
Description
Internal helper used by MizerSim rate methods. If time_range is missing,
all saved simulation times are selected; otherwise the request is delegated to
get_time_elements().
Usage
get_sim_rate_time_elements(sim, time_range)
Arguments
sim |
A |
time_range |
A numeric or character vector of times. |
Value
A named logical vector indicating the selected saved time steps.
Get size range array
Description
Helper function that returns an array (species x size) of logical values indicating whether that size bin is within the size limits specified by the arguments. Either the size limits can be the same for all species or they can be specified as vectors with one value for each species in the model.
Usage
get_size_range_array(
params,
min_w = min(params@w),
max_w = max(params@w),
min_l = NULL,
max_l = NULL,
...
)
Arguments
params |
MizerParams object |
min_w |
Smallest weight in size range. Defaults to smallest weight in the model. |
max_w |
Largest weight in size range. Defaults to largest weight in the model. |
min_l |
Smallest length in size range. If supplied, this takes
precedence over |
max_l |
Largest length in size range. If supplied, this takes precedence
over |
... |
Unused |
Value
A logical array (species x size), with dimnames sp and w.
Length to weight conversion
If min_l is specified there is no need to specify min_w and so on.
However, if a length is specified (minimum or maximum) then it is necessary
for the species parameter data.frame to include the parameters a and b
that determine the relation between length l and weight w by
w = a l^b.
It is possible to mix length and weight constraints, e.g. by supplying a minimum weight and a maximum length, but this must be done the same for all species. The default values are the minimum and maximum weights of the spectrum, i.e., the full range of the size spectrum is used.
Apply a species-by-size rate function over saved simulation times
Description
Internal helper used by MizerSim rate methods whose one-time result is an
ArraySpeciesBySize. The helper applies the supplied rate function to each
selected time slice, stacks the results, and restores the appropriate mizer
array class when dimensions have not been dropped.
Usage
get_species_size_rate_from_sim(
sim,
time_range,
drop,
rate_fun,
value_name,
units = NULL,
representation = "point"
)
Arguments
sim |
A |
time_range |
A numeric or character vector of times. |
drop |
If |
rate_fun |
A function accepting a single simulation slice as returned by
|
value_name |
Name of the value stored in the returned array. |
units |
Optional units of the value stored in the returned array. |
Value
A time x species x size array, possibly with dimensions dropped.
Apply a species rate function over saved simulation times
Description
Internal helper used by MizerSim rate methods whose one-time result is a
named vector with one value for each species.
Usage
get_species_time_rate_from_sim(
sim,
time_range,
rate_fun,
value_name,
units = NULL
)
Arguments
sim |
A |
time_range |
A numeric or character vector of times. |
rate_fun |
A function accepting a single simulation slice as returned by
|
value_name |
Name of the value stored in the returned array. |
units |
Optional units of the value stored in the returned array. |
Value
An ArrayTimeBySpecies object with dimensions time x species.
Calculate steady state abundance
Description
This function calculates the steady state abundance by solving the transport equation with given growth and mortality rates. It sets up a tri-diagonal system and solves it.
Usage
get_steady_state_n(
params,
g,
mu,
D,
N0,
max_iterations = 500,
tol = 1e-10,
relax = 0.3
)
Arguments
params |
A MizerParams object |
g |
A matrix of growth rates (species x size) |
mu |
A matrix of mortality rates (species x size) |
D |
A matrix of diffusion rates (species x size) |
N0 |
A vector with the abundance at the smallest size for each species |
max_iterations |
Maximum number of Picard iterations used when a flux limiter is active. |
tol |
Relative convergence tolerance for the Picard iteration. |
relax |
Under-relaxation factor in (0, 1] for the Picard iteration when a flux limiter is active. |
Details
The spatial discretisation of the advective flux is read from the
flux entry of the second_order_w slot of params. With a second-order
flux scheme active the steady state must match the one that project() converges
to. Because the limiter depends on the solution, the steady state is then
found by an under-relaxed Picard iteration: the limiter is frozen at the
current iterate, the resulting tridiagonal system is solved, and the iterate
is updated towards that solution, repeating until it converges. (At dt = 1
the limited operator is not diagonally dominant, so the plain fixed-point map
only stalls; under-relaxation makes it converge.)
The returned abundance is held at zero above each species' w_max, the same
upper boundary condition that project() imposes (via zero_above_support()
in project_n()). Without this the bottom-up solve would carry density above
w_max whenever growth is still positive there or diffusion pushes density
past it.
Value
A matrix with the steady state abundance
Get array indices for a time range in a MizerSim object
Description
Internal helper to select the saved time points whose times lie between the
smallest and largest values in time_range, inclusive.
Usage
get_time_elements(sim, time_range, slot_name = "n")
Arguments
sim |
A MizerSim object. |
time_range |
A numeric or character vector of times. Only the range of
values matters, so all saved times between |
slot_name |
Obsolete, kept only for backward compatibility with early versions where different time-based slots could have different time grids. Leave at the default. |
Value
A named logical vector, with one entry for each saved time in sim,
indicating whether that time lies in the requested range.
Description of indicator functions
Description
Mizer provides a range of functions to calculate indicators from a MizerSim or MizerParams object.
Details
When called with a MizerSim object, these functions return a time series
of values. When called with a MizerParams object, they return a single
value calculated from the initial abundances stored in the params object.
A list of available indicator functions is given in the table below
| Function | Returns | Description |
getProportionOfLargeFish() | A vector with values at each time step (or a single value for MizerParams). | Calculates the proportion of large fish through time. The threshold value can be specified. It is possible to calculation the proportion of large fish based on either length or weight. |
getMeanWeight() | A vector with values at each saved time step (or a single value for MizerParams). | The mean weight of the community through time. This is calculated as the total biomass of the community divided by the total abundance. |
getMeanMaxWeight() | Depends on the measure argument. If measure = “both” then you get a matrix with two columns, one with values by numbers, the other with values by biomass at each saved time step (or a named vector for MizerParams). If measure = “numbers” or “biomass” you get a vector of the respective values at each saved time step (or a single value for MizerParams). | The mean maximum weight of the community through time. This can be calculated by numbers or by biomass. See the help file for more details. |
getCommunitySlope() | A data.frame with four columns: time step, slope, intercept and the coefficient of determination (or a single-row data.frame for MizerParams). | Calculates the slope of the community abundance spectrum through time by performing a linear regression on the logged total numerical abundance and logged body size. |
See Also
summary_functions, plotting_functions
Initial values for fish spectra
Description
Values used as starting values for simulations with project().
Usage
initialN(params) <- value
initialN(object)
Arguments
params |
A MizerParams object |
value |
A matrix with dimensions species x size holding the initial number densities for the fish spectra. |
object |
An object of class MizerParams or MizerSim |
Value
An ArraySpeciesBySize object with dimensions species x size holding
the initial number densities for the fish spectra.
See Also
initialNResource(), initialNOther()
Examples
# Doubling abundance of Cod in the initial state of the North Sea model
params <- NS_params
initialN(params)["Cod", ] <- 2 * initialN(params)["Cod", ]
Initial values for other ecosystem components
Description
Values used as starting values for simulations with project().
Usage
initialNOther(params) <- value
initialNOther(object)
Arguments
params |
A MizerParams object |
value |
A named list with the initial values of other ecosystem components |
object |
An object of class MizerParams or MizerSim |
Value
A named list with the initial values of other ecosystem components
See Also
initialNResource(), initialN()
Other extension tools:
NOther(),
clearExtensionChain(),
coerceToExtensionClass(),
getRegisteredExtensions(),
recordExtension(),
registerExtension(),
registerExtensions(),
setComponent(),
setRateFunction()
Initial value for resource spectrum
Description
Value used as starting value for simulations with project().
Usage
initialNResource(params) <- value
initialNResource(object)
Arguments
params |
A MizerParams object |
value |
A vector with the initial number densities for the resource spectrum |
object |
An object of class MizerParams or MizerSim |
Value
A vector with the initial number densities for the resource spectrum
See Also
Examples
# Doubling resource abundance in the initial state of the North Sea model
params <- NS_params
initialNResource(params) <- 2 * initialNResource(params)
Extract the initial state from a simulation
Description
Returns the MizerParams object underlying the simulation with its initial
abundances set to the abundances at the initial time of the
simulation.
Usage
initialParams(sim)
Arguments
sim |
A |
Value
A MizerParams object with initial state of the simulation.
See Also
Examples
sim <- project(NS_params, t_max = 20, effort = 0.5)
params_start <- initialParams(sim)
Initial fishing effort
Description
The fishing effort is a named vector, specifying for each fishing gear the
effort invested into fishing with that gear. The effort value for each gear
is multiplied by the catchability and the selectivity to determine the
fishing mortality imposed by that gear, see setFishing() for more details.
The initial effort you have set can be overruled when running a simulation
by providing an effort argument to project() which allows you to
specify a time-varying effort.
Usage
initial_effort(params)
initial_effort(params) <- value
Arguments
params |
A MizerParams object |
value |
A vector or scalar with the initial fishing effort, see Details below. |
Details
A valid effort vector is a named vector with one effort value for each gear. However you can also supply the effort value in different ways:
a scalar, which is then replicated for each gear
an unnamed vector, which is then assumed to be in the same order as the gears in the params object
a named vector in which the gear names have a different order than in the params object. This is then sorted correctly.
a named vector which only supplies values for some of the gears. The effort for the other gears is then set to the default effort returned by
validEffortVector(), which depends on the defaults edition.
These conversions are done by the function validEffortVector().
An effort argument will lead to an error if it is either
unnamed and of the wrong length
named but where some names do not match any of the gears
not numeric
Value
A named effort vector ordered by gear.
Alias for NS_interaction
Description
An alias provided for backward compatibility with mizer version <= 2.3
Usage
inter
Format
A 12 x 12 matrix.
Source
Blanchard et al.
Test whether one extension chain is a suffix of another
Description
An empty candidate is always a suffix. Order and values must match
exactly for the overlapping tail.
Usage
isSuffixChain(candidate, chain)
Arguments
candidate |
Named character vector to test. |
chain |
Named character vector that may contain |
Value
TRUE if candidate is a suffix of chain, FALSE otherwise.
Test whether a requirement string is a dotted version number
Description
Test whether a requirement string is a dotted version number
Usage
isVersionRequirement(requirement)
Arguments
requirement |
Character string. |
Value
TRUE if requirement matches "X.Y.Z..." (digits and dots only).
Weight based knife-edge selectivity function
Description
A knife-edge selectivity function where weights greater or equal to
knife_edge_size are fully selected and no fish smaller than this size
are selected.
Usage
knife_edge(w, knife_edge_size, ...)
Arguments
w |
Vector of sizes. |
knife_edge_size |
The weight at which the knife-edge operates. |
... |
Unused |
Details
You would not usually call this function directly. Instead, set the sel_func
column in gear_params() to "knife_edge" and provide knife_edge_size as
an additional column. setFishing() will then call this function
automatically when calculating the selectivity array.
Value
Vector of selectivities at the given sizes.
See Also
gear_params() for setting the knife_edge_size parameter.
Other selectivity functions:
double_sigmoid_length(),
sigmoid_length(),
sigmoid_weight()
Examples
knife_edge(w = c(1, 10, 100, 1000), knife_edge_size = 100)
Length-weight conversion
Description
For each species, convert between length and weight using the relationship
w_i = a_i l_i^{b_i}
or
l_i = (w_i / a_i)^{1/b_i}
where a and b are taken from the species parameter data frame and
i is the species index.
Usage
l2w(l, species_params)
w2l(w, species_params)
Arguments
l |
Lengths in cm. Either a single number used for all species or a vector with one number for each species. |
species_params |
A species parameter data frame or a MizerParams object. |
w |
Weights in grams. Either a single number used for all species or a vector with one number for each species. |
Details
This is useful for converting a length-based species parameter to a weight-based species parameter.
Value
A vector with one entry for each species. l2w() returns a vector
of weights in grams and w2l() returns a vector of lengths in cm.
Helper function to produce nice breaks on logarithmic axes
Description
This is needed when the logarithmic y-axis spans less than one order of magnitude, in which case the ggplot2 default produces no ticks.
Usage
log_breaks(n = 6)
Arguments
n |
Approximate number of ticks |
Details
Thanks to Heather Turner at https://stackoverflow.com/questions/14255533/pretty-ticks-for-log-normal-scale-using-ggplot2-dynamic-not-manual
Value
A function that can be used as the break argument in calls to scale_y_continuous() or scale_x_continuous()
Lognormal predation kernel
Description
This is the most commonly-used predation kernel. The log of the predator/prey mass ratio is normally distributed.
Usage
lognormal_pred_kernel(ppmr, beta, sigma)
Arguments
ppmr |
A vector of predator/prey size ratios |
beta |
The preferred predator/prey size ratio |
sigma |
The width parameter of the log-normal kernel |
Details
Writing the predator mass as w and the prey mass as w_p,
the feeding kernel is given as
\phi_i(w, w_p) =
\exp \left[ \frac{-(\ln(w / w_p / \beta_i))^2}{2\sigma_i^2} \right]
if w/w_p is larger than 1 and zero otherwise. Here \beta_i is the
preferred predator-prey mass ratio and \sigma_i determines the width of
the kernel. These two parameters need to be given in the species parameter
dataframe in the columns beta and sigma.
This function is called from setPredKernel() to set up the
predation kernel slots in a MizerParams object.
Value
A vector giving the value of the predation kernel at each of the
predator/prey mass ratios in the ppmr argument.
See Also
Other predation kernel:
box_pred_kernel(),
power_law_pred_kernel(),
truncated_lognormal_pred_kernel()
Examples
params <- NS_params
plot(w_full(params), getPredKernel(params)["Cod", 10, ], type="l", log="x")
# The restriction that the kernel is zero for w/w_p < 1 is more
# noticeable for larger sigma
species_params(params)$sigma <- 4
plot(w_full(params), getPredKernel(params)["Cod", 10, ], type="l", log="x")
Build a versioned extension list from requirements and versions
Description
Build a versioned extension list from requirements and versions
Usage
makeExtensions(requirements, versions = character())
Arguments
requirements |
A named character vector of requirement strings. |
versions |
A named character vector of version stamps. Names not present
default to |
Value
A named list whose entries are
c(requirement = ..., version = ...), or an empty character vector when
requirements is empty.
Construct a named vector of line widths for a plot
Description
Helper used by the plotting functions to give highlighted species a thicker line than the rest.
Usage
make_linesize(levels, highlight)
Arguments
levels |
Character vector of the legend levels (usually species names). |
highlight |
Name or vector of names of the levels to be highlighted with a thicker line. |
Value
A named numeric vector of line widths, one for each entry in
levels, with highlighted entries set to a larger value.
Tag a ggplot object as a mizer plot
Description
Attaches the tooltip information to a ggplot object and adds the
"mizer_plot" class so that plotHover() knows which aesthetics to show.
Usage
make_mizer_plot(plot, tooltip)
Arguments
plot |
A ggplot object. |
tooltip |
Character vector of variable names to include in the plotly tooltip. |
Value
The plot object with the tooltip stored as an attribute and
"mizer_plot" prepended to its class.
Designate species as background species
Description
Marks the specified set of species as background species by setting the
is_background column in their species parameters to TRUE. Background
species are handled differently in plots (displayed in grey) and their
abundances can be automatically adjusted to keep the community close to the
Sheldon spectrum (see adjustBackgroundSpecies() in the mizerExperimental
package).
Usage
markBackground(object, species = NULL)
Arguments
object |
An object of class MizerParams or MizerSim. |
species |
The species to be selected. Optional. By default all target species are selected. A vector of species names, or a numeric vector with the species indices, or a logical vector indicating for each species whether it is to be selected (TRUE) or not. |
Value
An object of the same class as the object argument
See Also
Examples
params <- markBackground(NS_params,
species = c("Sprat", "Sandeel", "N.pout"))
any(species_params(params)$is_background)
Match biomasses to observations
Description
The function adjusts the abundances of the species in the model so that their
biomasses match with observations.
Usage
matchBiomasses(params, species = NULL, info_level = 3, ...)
Arguments
params |
A MizerParams object |
species |
The species to be affected. Optional. By default all observed biomasses will be matched. A vector of species names, or a numeric vector with the species indices, or a logical vector indicating for each species whether it is to be affected (TRUE) or not. |
info_level |
Controls the amount of information messages that are shown. Higher levels lead to more messages. |
... |
Additional arguments passed to the method. |
Details
The function works by multiplying for each species the abundance density
at all sizes by the same factor. This will of course not give a steady
state solution, even if the initial abundance densities were at steady state.
So after using this function you may want to use steady() to run the model
to steady state, after which of course the biomasses will no longer match
exactly. You could then iterate this process. This is described in the
blog post at https://blog.mizer.sizespectrum.org/posts/2021-08-20-a-5-step-recipe-for-tuning-the-model-steady-state/.
Before you can use this function you will need to have added a
biomass_observed column to your model which gives the observed biomass in
grams. For species for which you have no observed biomass, you should set
the value in the biomass_observed column to 0 or NA.
Biomass observations usually only include individuals above a certain size.
This size should be specified in a biomass_cutoff column of the species
parameter data frame. If this is missing, it is assumed that all sizes are
included in the observed biomass, i.e., it includes larval biomass.
Value
A MizerParams object
Examples
params <- NS_params
species_params(params)$biomass_observed <-
c(0.8, 61, 12, 35, 1.6, 20, 10, 7.6, 135, 60, 30, 78)
species_params(params)$biomass_cutoff <- 10
params <- calibrateBiomass(params)
params <- matchBiomasses(params)
plotBiomassObservedVsModel(params)
Adjust model to produce observed growth
Description
Scales the search volume, the maximum consumption rate, the metabolic rate
and the external encounter rate
all by the same factor in order to achieve a growth rate that allows
individuals to reach their maturity size by their maturity age while keeping
the feeding level and the critical feeding level unchanged. Then recalculates
the size spectra using steadySingleSpecies().
Usage
matchGrowth(params, species = NULL, keep = c("egg", "biomass", "number"), ...)
Arguments
params |
A MizerParams object |
species |
The species to be affected. Optional. By default all species for which growth information is available will be affected. A vector of species names, or a numeric vector with the species indices, or a logical vector indicating for each species whether it is to be affected (TRUE) or not. |
keep |
A string determining which quantity is to be kept constant. The choices are "egg" which keeps the egg density constant, "biomass" which keeps the total biomass of the species constant and "number" which keeps the total number of individuals constant. |
... |
Additional arguments passed to the method. |
Details
Maturity size and age are taken from the w_mat and age_mat columns in the
species_params data frame. If age_mat is missing, mizer calculates it from
the von Bertalanffy growth curve parameters using age_mat_vB(). If those
are not available either for a species, the growth rate for that species will
not be changed.
Value
A modified MizerParams object with rescaled search volume, maximum
consumption rate and metabolic rate and rescaled species parameters
gamma,h, ks and k.
Examples
# Rescale rates so all species reach maturity by their maturity age.
# The search volume gamma is adjusted to achieve the correct growth rate.
species_params(NS_params)["Cod", "gamma"]
params <- matchGrowth(NS_params)
species_params(params)["Cod", "gamma"]
age_mat(params)["Cod"]
Match numbers to observations
Description
The function adjusts the numbers of the species in the model so that their
numbers match with observations.
Usage
matchNumbers(params, species = NULL, info_level = 3, ...)
Arguments
params |
A MizerParams object |
species |
The species to be affected. Optional. By default all observed numbers will be matched. A vector of species names, or a numeric vector with the species indices, or a logical vector indicating for each species whether it is to be affected (TRUE) or not. |
info_level |
Controls the amount of information messages that are shown. Higher levels lead to more messages. |
... |
Additional arguments passed to the method. |
Details
The function works by multiplying for each species the number density
at all sizes by the same factor. This will of course not give a steady
state solution, even if the initial number densities were at steady state.
So after using this function you may want to use steady() to run the model
to steady state, after which of course the numbers will no longer match
exactly. You could then iterate this process. This is described in the
blog post at https://blog.mizer.sizespectrum.org/posts/2021-08-20-a-5-step-recipe-for-tuning-the-model-steady-state/.
Before you can use this function you will need to have added a
number_observed column to your model which gives the observed number of
individuals. For species for which you have no observed number, you should set
the value in the number_observed column to 0 or NA.
Number observations usually only include individuals above a certain size.
This size should be specified in a number_cutoff column of the species
parameter data frame. If this is missing, it is assumed that all sizes are
included in the observed number, i.e., it includes larval number.
Value
A MizerParams object
Examples
params <- NS_params
species_params(params)$number_observed <-
c(0.8, 61, 12, 35, 1.6, 20, 10, 7.6, 135, 60, 30, 78)
species_params(params)$number_cutoff <- 10
params <- calibrateNumber(params)
params <- matchNumbers(params)
Match yields to observations
Description
This function has been deprecated and will be removed in the future unless
you have a use case for it. If you do have a use case for it, please let the
developers know by creating an issue at
https://github.com/sizespectrum/mizer/issues.
Usage
matchYields(params, species = NULL, info_level = 3, ...)
Arguments
params |
A MizerParams object |
species |
The species to be affected. Optional. By default all observed yields will be matched. A vector of species names, or a numeric vector with the species indices, or a logical vector indicating for each species whether it is to be affected (TRUE) or not. |
info_level |
Controls the amount of information messages that are shown. Higher levels lead to more messages. |
... |
Additional arguments passed to the method. |
Details
If you want to match the yields to observations, you should use the matchYield() function from the mizerExperimental package instead, which adjusts the catchability to match the yield rather than by adjusting the biomass.
The function adjusts the abundances of the species in the model so that their yearly yields under the given fishing mortalities match with observations.
The function works by multiplying for each species the abundance density
at all sizes by the same factor. This will of course not give a steady
state solution, even if the initial abundance densities were at steady state.
So after using this function you may want to use steady() to run the model
to steady state, after which of course the yields will no longer match
exactly. You could then iterate this process. This is described in the
blog post at https://blog.mizer.sizespectrum.org/posts/2021-08-20-a-5-step-recipe-for-tuning-the-model-steady-state/.
Before you can use this function you will need to have added a
yield_observed column to your model which gives the observed yields in
grams per year. For species for which you have no observed biomass, you
should set the value in the yield_observed column to 0 or NA.
Value
A MizerParams object
Examples
params <- NS_params
species_params(params)$yield_observed <-
c(0.8, 61, 12, 35, 1.6, 20, 10, 7.6, 135, 60, 30, 78)
gear_params(params)$catchability <-
c(1.3, 0.065, 0.31, 0.18, 0.98, 0.24, 0.37, 0.46, 0.18, 0.30, 0.27, 0.39)
params <- calibrateYield(params)
params <- matchYields(params)
plotYieldObservedVsModel(params)
Calculate diffusion rate
Description
Calculates the diffusion rate D_i(w) (grams^2/year) for each species.
This diffusion rate has two components:
The diffusion due due to the variability in prey sizes. This is the diffusion term from the jump-growth equation.
Any externally specified diffusion, which is added via
setExtDiffusion()
You would not usually call this function directly but instead use
getDiffusion(), which then calls this function unless an alternative
diffusion rate function has been registered, see setRateFunction().
Usage
projectDiffusion(params, n, n_pp, n_other, t = 0, feeding_level, ...)
## S3 method for class 'MizerParams'
projectDiffusion(params, n, n_pp, n_other, t = 0, feeding_level, ...)
mizerDiffusion(params, n, n_pp, n_other, t = 0, feeding_level, ...)
Arguments
params |
A MizerParams object |
n |
A matrix of species abundances (species x size). |
n_pp |
A vector of the resource abundance by size |
n_other |
A list of abundances for other dynamical components |
t |
The time for which to do the calculation (Not used by standard mizer rate functions but useful for extensions.) |
feeding_level |
An array (species x size) with the feeding level. If not provided, it is calculated from the given abundances. |
... |
Unused |
Value
A two dimensional array (species x size) holding the diffusion rate.
Get energy rate available for growth needed to project standard mizer model
Description
Calculates the energy rate g_i(w) (grams/year) available by species and
size for growth after metabolism, movement and reproduction have been
accounted for. Used by project() for performing simulations.
You would not usually call this
function directly but instead use getEGrowth(), which then calls this
function unless an alternative function has been registered, see below.
Usage
projectEGrowth(params, n, n_pp, n_other, t = 0, e_repro, e, ...)
## S3 method for class 'MizerParams'
projectEGrowth(params, n, n_pp, n_other, t = 0, e_repro, e, ...)
mizerEGrowth(params, n, n_pp, n_other, t = 0, e_repro, e, ...)
Arguments
params |
A MizerParams object |
n |
A matrix of species abundances (species x size). |
n_pp |
A vector of the resource abundance by size |
n_other |
A list of abundances for other dynamical components of the ecosystem |
t |
The time for which to do the calculation (Not used by standard mizer rate functions but useful for extensions with time-dependent parameters.) |
e_repro |
The energy available for reproduction as calculated by
|
e |
The energy available for reproduction and growth as calculated by
|
... |
Unused |
Details
The growth rate is calculated as the difference between the energy available
for reproduction and growth (obtainable with getEReproAndGrowth()) and
the energy used for reproduction (obtainable with getERepro()), but is
set to 0 if the result would be negative.
Value
A two dimensional array (species x size) with the growth rates.
Your own growth rate function
By default getEGrowth() calls mizerEGrowth(). However you can
replace this with your own alternative growth rate function. If
your function is called "myEGrowth" then you register it in a MizerParams
object params with
params <- setRateFunction(params, "EGrowth", "myEGrowth")
Your function will then be called instead of mizerEGrowth(), with the
same arguments.
See Also
Other mizer rate functions:
mizerERepro(),
mizerEReproAndGrowth(),
mizerEncounter(),
mizerFMort(),
mizerFMortGear(),
mizerFeedingLevel(),
mizerMort(),
mizerPredMort(),
mizerPredRate(),
mizerRDI(),
mizerRates(),
mizerResourceMort()
Get energy rate available for reproduction needed to project standard mizer model
Description
Calculates the energy rate (grams/year) available for reproduction after
growth and metabolism have been accounted for.
You would not usually call this
function directly but instead use getERepro(), which then calls this
function unless an alternative function has been registered, see below.
Usage
projectERepro(params, n, n_pp, n_other, t = 0, e, ...)
## S3 method for class 'MizerParams'
projectERepro(params, n, n_pp, n_other, t = 0, e, ...)
mizerERepro(params, n, n_pp, n_other, t = 0, e, ...)
Arguments
params |
A MizerParams object |
n |
A matrix of species abundances (species x size). |
n_pp |
A vector of the resource abundance by size |
n_other |
A list of abundances for other dynamical components of the ecosystem |
t |
The time for which to do the calculation (Not used by standard mizer rate functions but useful for extensions with time-dependent parameters.) |
e |
A two dimensional array (species x size) holding the energy available
for reproduction and growth as calculated by |
... |
Unused |
Value
A two dimensional array (species x size) holding
\psi_i(w)\max(0, E_{r.i}(w))
where E_{r.i}(w) is the rate at which energy becomes available for
growth and reproduction, calculated with mizerEReproAndGrowth(),
and \psi_i(w) is the proportion of this energy that is used for
reproduction. Negative entries in e are clipped to 0 before multiplying by
\psi_i(w). This proportion is taken from the params object and is set
with setReproduction().
Your own reproduction rate function
By default getERepro() calls mizerERepro(). However you can
replace this with your own alternative reproduction rate function. If
your function is called "myERepro" then you register it in a MizerParams
object params with
params <- setRateFunction(params, "ERepro", "myERepro")
Your function will then be called instead of mizerERepro(), with the
same arguments.
See Also
Other mizer rate functions:
mizerEGrowth(),
mizerEReproAndGrowth(),
mizerEncounter(),
mizerFMort(),
mizerFMortGear(),
mizerFeedingLevel(),
mizerMort(),
mizerPredMort(),
mizerPredRate(),
mizerRDI(),
mizerRates(),
mizerResourceMort()
Get energy rate available for reproduction and growth needed to project standard mizer model
Description
Calculates the energy rate
E_{r.i}(w) (grams/year) available to an
individual of species i and size w for reproduction and growth after
metabolism and movement have been accounted for.
You would not usually call this function directly but instead use
getEReproAndGrowth(), which then calls this function unless an alternative
function has been registered, see below.
Usage
projectEReproAndGrowth(
params,
n,
n_pp,
n_other,
t = 0,
encounter,
feeding_level,
...
)
## S3 method for class 'MizerParams'
projectEReproAndGrowth(
params,
n,
n_pp,
n_other,
t = 0,
encounter,
feeding_level,
...
)
mizerEReproAndGrowth(
params,
n,
n_pp,
n_other,
t = 0,
encounter,
feeding_level,
...
)
Arguments
params |
A MizerParams object |
n |
A matrix of species abundances (species x size). |
n_pp |
A vector of the resource abundance by size |
n_other |
A list of abundances for other dynamical components of the ecosystem |
t |
The time for which to do the calculation (Not used by standard mizer rate functions but useful for extensions with time-dependent parameters.) |
encounter |
An array (species x size) with the encounter rate as
calculated by |
feeding_level |
An array (species x size) with the feeding level as
calculated by |
... |
Unused |
Value
A two dimensional array (species x size) holding
E_{r.i}(w) = \alpha_i\, (1 - {\tt feeding\_level}_i(w))\,
{\tt encounter}_i(w) - {\tt metab}_i(w).
Due to the form of the feeding level, calculated by
getFeedingLevel(), if the feeding level is nonzero this can also be expressed as
E_{r.i}(w) = \alpha_i\, {\tt feeding\_level}_i(w)\,
h_i(w) - {\tt metab}_i(w)
where h_i is the maximum intake rate, set with
setMaxIntakeRate(). However this function is using the first equation
above so that it works also when the maximum intake rate is infinite, i.e.,
there is no satiation.
The assimilation rate \alpha_i is taken from the species parameter
data frame in params. The metabolic rate metab is taken from
params and set with setMetabolicRate().
The return value can be negative, which means that the energy intake does not cover the cost of metabolism and movement.
Your own energy rate function
By default getEReproAndGrowth() calls mizerEReproAndGrowth(). However you
can replace this with your own alternative energy rate function. If
your function is called "myEReproAndGrowth" then you register it in a
MizerParams object params with
params <- setRateFunction(params, "EReproAndGrowth", "myEReproAndGrowth")
Your function will then be called instead of mizerEReproAndGrowth(), with
the same arguments.
See Also
Other mizer rate functions:
mizerEGrowth(),
mizerERepro(),
mizerEncounter(),
mizerFMort(),
mizerFMortGear(),
mizerFeedingLevel(),
mizerMort(),
mizerPredMort(),
mizerPredRate(),
mizerRDI(),
mizerRates(),
mizerResourceMort()
Get encounter rate during projection
Description
Calculates the rate E_i(w) at which a predator of species i and
weight w encounters food (grams/year). You would not usually call this
function directly but instead use getEncounter(), which then calls this
function unless an alternative function has been registered, see below.
Usage
projectEncounter(params, n, n_pp, n_other, t = 0, ...)
## S3 method for class 'MizerParams'
projectEncounter(params, n, n_pp, n_other, t = 0, ...)
mizerEncounter(params, n, n_pp, n_other, t = 0, ...)
Arguments
params |
A MizerParams object |
n |
A matrix of species abundances (species x size). |
n_pp |
A vector of the resource abundance by size |
n_other |
A list of abundances for other dynamical components of the ecosystem |
t |
The time for which to do the calculation (Not used by standard mizer rate functions but useful for extensions with time-dependent parameters.) |
... |
Unused |
Value
A named two dimensional array (predator species x predator size) with the encounter rates.
Predation encounter
The encounter rate E_i(w) at which a predator of species i
and weight w encounters food has contributions from the encounter of
fish prey and of resource. This is determined by summing over all prey
species and the resource spectrum and then integrating over all prey sizes
w_p, weighted by predation kernel \phi(w,w_p):
E_i(w) = \gamma_i(w) \int
\left( \theta_{ip} N_R(w_p) + \sum_{j} \theta_{ij} N_j(w_p) \right)
\phi_i(w,w_p) w_p \, dw_p.
Here N_j(w) is the abundance density of species j and
N_R(w) is the abundance density of resource.
The overall prefactor \gamma_i(w) determines the predation power of the
predator. It could be interpreted as a search volume and is set with the
setSearchVolume() function. The predation kernel
\phi(w,w_p) is set with the setPredKernel() function. The
species interaction matrix \theta_{ij} is set with setInteraction()
and the resource interaction vector \theta_{ip} is taken from the
interaction_resource column in params@species_params.
Details
The encounter rate is multiplied by 1-f_0 to obtain the consumption
rate, where f_0 is the feeding level calculated with
getFeedingLevel(). This is used by the project() function for performing
simulations.
The function returns values also for sizes outside the size-range of the species. These values should not be used, as they are meaningless.
If your model contains additional components that you added with
setComponent() and for which you specified an encounter_fun function then
the encounters of these components will be included in the returned value.
Extension hook
projectEncounter() is the S3 generic used by extension-aware projections.
Extension packages can add methods for their marker classes and call
NextMethod() to compose encounter-rate changes. The MizerParams method
contains the standard mizer calculation and is also exported as
mizerEncounter() for compatibility.
Your own encounter function
By default getEncounter() calls mizerEncounter() on models without
extensions. However you can replace this with your own alternative encounter
function. If your function is called "myEncounter" then you register it in
a MizerParams object params with
params <- setRateFunction(params, "Encounter", "myEncounter")
Your function will then be called instead of mizerEncounter(), with the
same arguments.
See Also
Other mizer rate functions:
mizerEGrowth(),
mizerERepro(),
mizerEReproAndGrowth(),
mizerFMort(),
mizerFMortGear(),
mizerFeedingLevel(),
mizerMort(),
mizerPredMort(),
mizerPredRate(),
mizerRDI(),
mizerRates(),
mizerResourceMort()
Get the total fishing mortality rate from all fishing gears
Description
Calculates the total fishing mortality (in units 1/year) from all gears by
species and size.
The total fishing mortality is just the sum of the fishing mortalities
imposed by each gear, \mu_{f.i}(w)=\sum_g F_{g,i,w}.
You would not usually call this
function directly but instead use getFMort(), which then calls this
function unless an alternative function has been registered, see below.
Usage
projectFMort(params, n, n_pp, n_other, t = 0, effort, e_growth, pred_mort, ...)
## S3 method for class 'MizerParams'
projectFMort(params, n, n_pp, n_other, t = 0, effort, e_growth, pred_mort, ...)
mizerFMort(params, n, n_pp, n_other, t = 0, effort, e_growth, pred_mort, ...)
Arguments
params |
A MizerParams object |
n |
A matrix of species abundances (species x size). |
n_pp |
A vector of the resource abundance by size |
n_other |
A list of abundances for other dynamical components of the ecosystem |
t |
The time for which to do the calculation (Not used by standard mizer rate functions but useful for extensions with time-dependent parameters.) |
effort |
A vector with the effort for each fishing gear. |
e_growth |
An array (species x size) with the energy available for
growth as calculated by |
pred_mort |
A two dimensional array (species x size) with the predation
mortality as calculated by |
... |
Unused |
Value
An array (species x size) with the fishing mortality.
Your own fishing mortality function
By default getFMort() calls mizerFMort(). However you can
replace this with your own alternative fishing mortality function. If
your function is called "myFMort" then you register it in a MizerParams
object params with
params <- setRateFunction(params, "FMort", "myFMort")
Your function will then be called instead of mizerFMort(), with the
same arguments.
Note
Here: fishing mortality = catchability x selectivity x effort.
See Also
Other mizer rate functions:
mizerEGrowth(),
mizerERepro(),
mizerEReproAndGrowth(),
mizerEncounter(),
mizerFMortGear(),
mizerFeedingLevel(),
mizerMort(),
mizerPredMort(),
mizerPredRate(),
mizerRDI(),
mizerRates(),
mizerResourceMort()
Get the fishing mortality needed to project standard mizer model
Description
Calculates the fishing mortality rate F_{g,i,w} by gear, species and
size.
This is a helper function for mizerFMort().
Usage
mizerFMortGear(params, effort)
Arguments
params |
A MizerParams object |
effort |
A vector with the effort for each fishing gear. |
Value
A three dimensional array (gear x species x size) with the fishing mortality.
Note
Here: fishing mortality = catchability x selectivity x effort.
See Also
Other mizer rate functions:
mizerEGrowth(),
mizerERepro(),
mizerEReproAndGrowth(),
mizerEncounter(),
mizerFMort(),
mizerFeedingLevel(),
mizerMort(),
mizerPredMort(),
mizerPredRate(),
mizerRDI(),
mizerRates(),
mizerResourceMort()
Get feeding level needed to project standard mizer model
Description
You would not usually call this function directly but instead use
getFeedingLevel(), which then calls this function unless an alternative
function has been registered, see below.
Usage
projectFeedingLevel(params, n, n_pp, n_other, t = 0, encounter, ...)
## S3 method for class 'MizerParams'
projectFeedingLevel(params, n, n_pp, n_other, t = 0, encounter, ...)
mizerFeedingLevel(params, n, n_pp, n_other, t = 0, encounter, ...)
Arguments
params |
A MizerParams object |
n |
A matrix of species abundances (species x size). |
n_pp |
A vector of the resource abundance by size |
n_other |
A list of abundances for other dynamical components of the ecosystem |
t |
The time for which to do the calculation (Not used by standard mizer rate functions but useful for extensions with time-dependent parameters.) |
encounter |
A two dimensional array (predator species x predator size) with the encounter rate. |
... |
Unused |
Value
A two dimensional array (predator species x predator size) with the feeding level.
Feeding level
The feeding level f_i(w) is the
proportion of its maximum intake rate at which the predator is actually
taking in fish. It is calculated from the encounter rate E_i and the
maximum intake rate h_i(w) as
f_i(w) = \frac{E_i(w)}{E_i(w)+h_i(w)}.
The encounter rate E_i is passed as an argument or calculated with
getEncounter(). The maximum intake rate h_i(w) is
taken from the params object, and is set with
setMaxIntakeRate().
As a consequence of the above expression for the feeding level,
1-f_i(w) is the proportion of the food available to it that the
predator actually consumes.
Your own feeding level function
By default getFeedingLevel() calls mizerFeedingLevel(). However you can
replace this with your own alternative feeding level function. If
your function is called "myFeedingLevel" then you register it in a MizerParams
object params with
params <- setRateFunction(params, "FeedingLevel", "myFeedingLevel")
Your function will then be called instead of mizerFeedingLevel(), with the
same arguments.
See Also
The feeding level is used in mizerEReproAndGrowth() and in
mizerPredRate().
Other mizer rate functions:
mizerEGrowth(),
mizerERepro(),
mizerEReproAndGrowth(),
mizerEncounter(),
mizerFMort(),
mizerFMortGear(),
mizerMort(),
mizerPredMort(),
mizerPredRate(),
mizerRDI(),
mizerRates(),
mizerResourceMort()
Get total mortality rate needed to project standard mizer model
Description
Calculates the total mortality rate \mu_i(w) (in units 1/year) on each
species by size from predation mortality, background mortality and fishing
mortality.
You would not usually call this
function directly but instead use getMort(), which then calls this
function unless an alternative function has been registered, see below.
Usage
projectMort(params, n, n_pp, n_other, t = 0, f_mort, pred_mort, ...)
## S3 method for class 'MizerParams'
projectMort(params, n, n_pp, n_other, t = 0, f_mort, pred_mort, ...)
mizerMort(params, n, n_pp, n_other, t = 0, f_mort, pred_mort, ...)
Arguments
params |
A MizerParams object |
n |
A matrix of species abundances (species x size). |
n_pp |
A vector of the resource abundance by size |
n_other |
A list of abundances for other dynamical components of the ecosystem |
t |
The time for which to do the calculation (Not used by standard mizer rate functions but useful for extensions with time-dependent parameters.) |
f_mort |
A two dimensional array (species x size) with the fishing mortality |
pred_mort |
A two dimensional array (species x size) with the predation mortality |
... |
Unused |
Details
If your model contains additional components that you added with
setComponent() and for which you specified a mort_fun function then
the mortality inflicted by these components will be included in the returned
value.
Value
A named two dimensional array (species x size) with the total mortality rates.
Your own mortality function
By default getMort() calls mizerMort(). However you can
replace this with your own alternative mortality function. If
your function is called "myMort" then you register it in a MizerParams
object params with
params <- setRateFunction(params, "Mort", "myMort")
Your function will then be called instead of mizerMort(), with the
same arguments.
See Also
Other mizer rate functions:
mizerEGrowth(),
mizerERepro(),
mizerEReproAndGrowth(),
mizerEncounter(),
mizerFMort(),
mizerFMortGear(),
mizerFeedingLevel(),
mizerPredMort(),
mizerPredRate(),
mizerRDI(),
mizerRates(),
mizerResourceMort()
Get total predation mortality rate needed to project standard mizer model
Description
Calculates the total predation mortality rate \mu_{p,i}(w_p) (in units
of 1/year) on each prey species by prey size:
\mu_{p.i}(w_p) = \sum_j {\tt pred\_rate}_j(w_p)\, \theta_{ji}.
You would not usually call this
function directly but instead use getPredMort(), which then calls this
function unless an alternative function has been registered, see below.
Usage
projectPredMort(params, n, n_pp, n_other, t = 0, pred_rate, ...)
## S3 method for class 'MizerParams'
projectPredMort(params, n, n_pp, n_other, t = 0, pred_rate, ...)
mizerPredMort(params, n, n_pp, n_other, t = 0, pred_rate, ...)
Arguments
params |
A MizerParams object |
n |
A matrix of species abundances (species x size). |
n_pp |
A vector of the resource abundance by size |
n_other |
A list of abundances for other dynamical components of the ecosystem |
t |
The time for which to do the calculation (Not used by standard mizer rate functions but useful for extensions with time-dependent parameters.) |
pred_rate |
A two dimensional array (predator species x prey size) with the predation rate, where prey size runs over fish community plus resource spectrum. |
... |
Unused |
Value
A two dimensional array (prey species x prey size) with the predation mortality
Your own predation mortality function
By default getPredMort() calls mizerPredMort(). However you can
replace this with your own alternative predation mortality function. If
your function is called "myPredMort" then you register it in a MizerParams
object params with
params <- setRateFunction(params, "PredMort", "myPredMort")
Your function will then be called instead of mizerPredMort(), with the
same arguments.
See Also
Other mizer rate functions:
mizerEGrowth(),
mizerERepro(),
mizerEReproAndGrowth(),
mizerEncounter(),
mizerFMort(),
mizerFMortGear(),
mizerFeedingLevel(),
mizerMort(),
mizerPredRate(),
mizerRDI(),
mizerRates(),
mizerResourceMort()
Get predation rate needed to project standard mizer model
Description
Calculates the potential rate (in units 1/year) at which a prey individual of
a given size w is killed by predators from species j. In formulas
{\tt pred\_rate}_j(w_p) = \int \phi_j(w,w_p) (1-f_j(w))
\gamma_j(w) N_j(w) \, dw.
This potential rate is used in the function mizerPredMort() to
calculate the realised predation mortality rate on the prey individual.
You would not usually call this
function directly but instead use getPredRate(), which then calls this
function unless an alternative function has been registered, see below.
Usage
projectPredRate(params, n, n_pp, n_other, t = 0, feeding_level, ...)
## S3 method for class 'MizerParams'
projectPredRate(params, n, n_pp, n_other, t = 0, feeding_level, ...)
mizerPredRate(params, n, n_pp, n_other, t = 0, feeding_level, ...)
Arguments
params |
A MizerParams object |
n |
A matrix of species abundances (species x size). |
n_pp |
A vector of the resource abundance by size |
n_other |
A list of abundances for other dynamical components of the ecosystem |
t |
The time for which to do the calculation (Not used by standard mizer rate functions but useful for extensions with time-dependent parameters.) |
feeding_level |
An array (species x size) with the feeding level as
calculated by |
... |
Unused |
Value
A named two dimensional array (predator species x prey size) with the predation rate, where the prey size runs over fish community plus resource spectrum.
Your own predation rate function
By default getPredRate() calls mizerPredRate(). However you can
replace this with your own alternative predation rate function. If
your function is called "myPredRate" then you register it in a MizerParams
object params with
params <- setRateFunction(params, "PredRate", "myPredRate")
Your function will then be called instead of mizerPredRate(), with
the same arguments.
See Also
Other mizer rate functions:
mizerEGrowth(),
mizerERepro(),
mizerEReproAndGrowth(),
mizerEncounter(),
mizerFMort(),
mizerFMortGear(),
mizerFeedingLevel(),
mizerMort(),
mizerPredMort(),
mizerRDI(),
mizerRates(),
mizerResourceMort()
Get density-independent rate of reproduction needed to project standard mizer model
Description
Calculates the density-independent rate of total egg production
R_{di} (units 1/year) before density dependence, by species.
You would not usually call this
function directly but instead use getRDI(), which then calls this
function unless an alternative function has been registered, see below.
Usage
projectRDI(
params,
n,
n_pp,
n_other,
t = 0,
e_growth,
mort,
e_repro,
diffusion = NULL,
...
)
## S3 method for class 'MizerParams'
projectRDI(
params,
n,
n_pp,
n_other,
t = 0,
e_growth,
mort,
e_repro,
diffusion = NULL,
...
)
mizerRDI(
params,
n,
n_pp,
n_other,
t = 0,
e_growth,
mort,
e_repro,
diffusion = NULL,
...
)
Arguments
params |
A MizerParams object |
n |
A matrix of species abundances (species x size). |
n_pp |
A vector of the resource abundance by size |
n_other |
A list of abundances for other dynamical components of the ecosystem |
t |
The time for which to do the calculation (Not used by standard mizer rate functions but useful for extensions with time-dependent parameters.) |
e_growth |
An array (species x size) with the energy available for
growth as calculated by |
mort |
An array (species x size) with the mortality rate as calculated
by |
e_repro |
An array (species x size) with the energy available for
reproduction as calculated by |
diffusion |
An array (species x size) with the diffusion rate as
calculated by |
... |
Unused |
Details
This rate is obtained by taking the per capita rate E_r(w)\psi(w) at
which energy is invested in reproduction, as calculated by getERepro(),
multiplying it by the number of individualsN(w) and integrating over
all sizes w and then multiplying by the reproductive efficiency
\epsilon and dividing by the egg size w_min, and by a factor of two
to account for the two sexes:
R_{di} = \frac{\epsilon}{2 w_{min}} \int N(w) E_r(w) \psi(w) \, dw
Used by getRDD() to calculate the actual, density dependent rate.
See setReproduction() for more details.
Value
A numeric vector with the rate of egg production for each species.
Your own reproduction function
By default getRDI() calls mizerRDI(). However you can
replace this with your own alternative reproduction function. If
your function is called "myRDI" then you register it in a MizerParams
object params with
params <- setRateFunction(params, "RDI", "myRDI")
Your function will then be called instead of mizerRDI(), with the
same arguments. For an example of an alternative reproduction function
see constantEggRDI().
See Also
Other mizer rate functions:
mizerEGrowth(),
mizerERepro(),
mizerEReproAndGrowth(),
mizerEncounter(),
mizerFMort(),
mizerFMortGear(),
mizerFeedingLevel(),
mizerMort(),
mizerPredMort(),
mizerPredRate(),
mizerRates(),
mizerResourceMort()
Get all rates needed to project standard mizer model
Description
Calls other rate functions in sequence and collects the results in a list.
projectRates() is an S3 generic used by extension-aware
projections to calculate all rates. Models without extensions keep using
mizerRates() directly. The base method mirrors mizerRates() but calls
migrated projection hooks directly, starting with projectEncounter().
Usage
mizerRates(params, n, n_pp, n_other, t = 0, effort, rates_fns, ...)
projectRates(params, n, n_pp, n_other, t = 0, effort, rates_fns, ...)
Arguments
params |
A MizerParams object |
n |
A matrix of species abundances (species x size). |
n_pp |
A vector of the resource abundance by size |
n_other |
A list of abundances for other dynamical components of the ecosystem |
t |
The time for which to do the calculation (Not used by standard mizer rate functions but useful for extensions with time-dependent parameters.) |
effort |
The effort for each fishing gear |
rates_fns |
Named list of the functions to call to calculate the rates. Note that this list holds the functions themselves, not their names. |
... |
Unused |
Details
By default this function returns a list with the following components:
encounter from
mizerEncounter()feeding_level from
mizerFeedingLevel()e from
mizerEReproAndGrowth()e_repro from
mizerERepro()e_growth from
mizerEGrowth()pred_rate from
mizerPredRate()pred_mort from
mizerPredMort()f_mort from
mizerFMort()mort from
mizerMort()rdi from
mizerRDI()rdd from
BevertonHoltRDD()resource_mort from
mizerResourceMort()
However you can replace any of these rate functions by your own rate
function if you wish, see setRateFunction() for details.
Value
List of rates.
See Also
Other mizer rate functions:
mizerEGrowth(),
mizerERepro(),
mizerEReproAndGrowth(),
mizerEncounter(),
mizerFMort(),
mizerFMortGear(),
mizerFeedingLevel(),
mizerMort(),
mizerPredMort(),
mizerPredRate(),
mizerRDI(),
mizerResourceMort()
Get predation mortality rate for resource needed to project standard mizer model
Description
Calculates the predation mortality rate \mu_p(w) on the resource
spectrum by resource size (in units 1/year).
You would not usually call this
function directly but instead use getResourceMort(), which then calls this
function unless an alternative function has been registered, see below.
Usage
projectResourceMort(params, n, n_pp, n_other, t = 0, pred_rate, ...)
## S3 method for class 'MizerParams'
projectResourceMort(params, n, n_pp, n_other, t = 0, pred_rate, ...)
mizerResourceMort(params, n, n_pp, n_other, t = 0, pred_rate, ...)
Arguments
params |
A MizerParams object |
n |
A matrix of species abundances (species x size). |
n_pp |
A vector of the resource abundance by size |
n_other |
A list of abundances for other dynamical components of the ecosystem |
t |
The time for which to do the calculation (Not used by standard mizer rate functions but useful for extensions with time-dependent parameters.) |
pred_rate |
A two dimensional array (predator species x prey size) with the predation rate, where the prey size runs over fish community plus resource spectrum. |
... |
Unused |
Value
A vector of mortality rate by resource size.
Your own resource mortality function
By default getResourceMort() calls mizerResourceMort(). However you can
replace this with your own alternative resource mortality function. If
your function is called "myResourceMort" then you register it in a MizerParams
object params with
params <- setRateFunction(params, "ResourceMort", "myResourceMort")
Your function will then be called instead of mizerResourceMort(), with the
same arguments.
See Also
Other mizer rate functions:
mizerEGrowth(),
mizerERepro(),
mizerEReproAndGrowth(),
mizerEncounter(),
mizerFMort(),
mizerFMortGear(),
mizerFeedingLevel(),
mizerMort(),
mizerPredMort(),
mizerPredRate(),
mizerRDI(),
mizerRates()
Whether the core mizer slots of an object need upgrading
Description
Whether the core mizer slots of an object need upgrading
Usage
mizer_needs_upgrading(params)
Arguments
params |
A MizerParams object. |
Value
TRUE or FALSE.
Calculate a selected subset of the rates
Description
Internal helper used by the MizerSim rate getters. Given rate functions
already resolved once with projectRateFunctions(), it calculates only those
rates needed to obtain the requested targets (plus their dependencies),
avoiding both the per-time-step cost of re-resolving the functions and the
cost of computing rates that are not required. The individual calculations
mirror those in mizerRates().
Usage
mizer_rates_subset(
params,
n,
n_pp,
n_other,
t,
effort,
rates_fns,
targets,
...
)
Arguments
params |
A valid |
n |
A matrix of species abundances (species x size). |
n_pp |
A vector of the resource abundance by size. |
n_other |
A named list of the abundances of other components. |
t |
The time for the calculation. |
effort |
The fishing effort. Only used when a target requires the fishing mortality. |
rates_fns |
Named list of resolved rate functions, as returned by
|
targets |
Character vector of rate names (as in |
... |
Passed on to the individual rate functions. |
Value
A named list of the calculated rates, using the same element names
as the list returned by mizerRates().
Determine the tooltip variables for a mizer plot
Description
Works out which variables should appear in the plotly tooltip, including the legend variable only when it differs from the grouping variable, plus any extra variables.
Usage
mizer_tooltip_vars(
frame,
group_var,
x_var,
y_var,
legend_var = NULL,
extra = NULL
)
Arguments
frame |
The data frame underlying the plot. |
group_var |
Name of the grouping variable. |
x_var |
Name of the variable on the x-axis. |
y_var |
Name of the variable on the y-axis. |
legend_var |
Optional name of the legend variable. |
extra |
Optional character vector of additional variable names to include. |
Value
A character vector of variable names for the tooltip.
Determine which rates must be calculated to obtain a set of target rates
Description
Internal helper returning the transitive closure of targets over
.rate_dependencies, in an order in which the rates can be calculated (each
rate appears after all the rates it depends on).
Usage
needed_rates(targets)
Arguments
targets |
Character vector of rate names (as in |
Value
A character vector of rate names.
Determine whether a MizerParams or MizerSim object needs to be upgraded
Description
Looks at the mizer version that was used to last update the object and
returns TRUE if changes since that version require an upgrade of the object.
You would not usually have to call this function. Upgrades are initiated
automatically by validParams and validSim when necessary.
Usage
needs_upgrading(object)
Arguments
object |
A MizerParams or MizerSim object |
Value
TRUE or FALSE
Set up parameters for a community-type model
Description
This functions creates a MizerParams object describing a
community-type model.
The function has many arguments, all of which have default values.
Usage
newCommunityParams(
max_w = 1e+06,
min_w = 0.001,
no_w = 100,
min_w_pp = 1e-10,
z0 = 0.1,
alpha = 0.2,
f0 = 0.7,
h = 10,
gamma = NA,
beta = 100,
sigma = 2,
n = 2/3,
kappa = 1000,
lambda = 2.05,
r_pp = 10,
knife_edge_size = 1000,
reproduction,
second_order_w = FALSE,
info_level = 2
)
Arguments
max_w |
The maximum size of the community. The |
min_w |
The minimum size of the community. |
no_w |
The number of size bins in the consumer spectrum. |
min_w_pp |
The smallest size of the resource spectrum. By default this is set to the smallest value at which any of the consumers can feed. |
z0 |
The background mortality of the community. |
alpha |
The assimilation efficiency of the community. |
f0 |
The average feeding level of individuals who feed on a power-law
spectrum. This value is used to calculate the search rate parameter
|
h |
The coefficient of the maximum food intake rate. |
gamma |
Volumetric search rate. Passed through to
|
beta |
The preferred predator prey mass ratio. |
sigma |
The width of the prey preference. |
n |
The allometric growth exponent. Used as allometric exponent for the maximum intake rate of the community as well as the intrinsic growth rate of the resource. |
kappa |
The coefficient |
lambda |
Used to set power-law exponent for resource capacity if the
|
r_pp |
Growth rate parameter for the resource spectrum. |
knife_edge_size |
The size at the edge of the knife-edge-selectivity function. |
reproduction |
The constant reproduction in the smallest size class of the community spectrum. By default this is set to the rate required to maintain the constructed initial egg abundance. |
second_order_w |
|
info_level |
Controls the amount of information messages that are shown when the function sets default values for parameters. Higher levels lead to more messages. |
Details
A community model has several features that distinguish it from a multi-species model:
Species identities of individuals are ignored. All are aggregated into a single community.
The resource spectrum only extends to the start of the community spectrum.
Reproductive rate is constant, independent of the energy invested in reproduction, which is set to 0.
Standard metabolism is turned off (the parameter
ksis set to 0). Consequently, the growth rate is now determined solely by the assimilated food
Fishing selectivity is modelled as a knife-edge function with one parameter,
knife_edge_size, which determines the size at which species are
selected.
Because this constructor does not yet set up stochastic growth by diffusion,
the size grid is not extended beyond the community's maximum size max_w
(so that w_max = w_repro_max), rather than leaving the headroom that
newMultispeciesParams() uses to accommodate stochastic growth. This will be
revisited once these constructors gain a diffusion parameter, see
https://github.com/sizespectrum/mizer/issues/339.
The resulting MizerParams object can be projected forward using
project() like any other MizerParams object. When projecting
the community model it may be necessary to keep a small time step size
dt of around 0.1 to avoid any instabilities with the solver. You can
check for these numerical instabilities by plotting the biomass or abundance
through time after the projection.
Value
An object of type MizerParams
References
K. H. Andersen,J. E. Beyer and P. Lundberg, 2009, Trophic and individual efficiencies of size-structured communities, Proceedings of the Royal Society, 276, 109-114
See Also
Other functions for setting up models:
newMultispeciesParams(),
newSingleSpeciesParams(),
newTraitParams()
Examples
params <- newCommunityParams()
sim <- project(params, t_max = 10)
plotBiomass(sim)
plotSpectra(sim, power = 2)
# More satiation. More mortality
params <- newCommunityParams(f0 = 0.8, z0 = 0.4)
sim <- project(params, t_max = 10)
plotBiomass(sim)
plotSpectra(sim, power = 2)
Set up parameters for a general multispecies model
Description
Sets up a multi-species size spectrum model by filling all slots in the
MizerParams object based on user-provided or default
parameters. There is a long list of arguments, but almost
all of them have sensible default values. The only required argument is
the species_params data frame. All arguments are described in more
details in the sections below the list.
Usage
newMultispeciesParams(
species_params,
interaction = NULL,
no_w = 100,
min_w = 0.001,
max_w = NA,
min_w_pp = NA,
pred_kernel = NULL,
search_vol = NULL,
intake_max = NULL,
metab = NULL,
p = 0.7,
ext_mort = NULL,
z0pre = 0.6,
z0exp = n - 1,
ext_encounter = NULL,
maturity = NULL,
repro_prop = NULL,
RDD = "BevertonHoltRDD",
kappa = 1e+11,
n = 2/3,
resource_rate = 10,
resource_capacity = kappa,
lambda = 2.05,
w_pp_cutoff = 10,
resource_dynamics = "resource_semichemostat",
gear_params = NULL,
selectivity = NULL,
catchability = NULL,
initial_effort = NULL,
second_order_w = FALSE,
info_level = 3,
z0 = deprecated(),
r_pp = deprecated()
)
Arguments
species_params |
A data frame of species-specific parameter values. |
interaction |
Optional interaction matrix of the species (predator species x prey species). By default all entries are 1. See "Setting interaction matrix" section below. |
no_w |
The number of size bins in the consumer spectrum. |
min_w |
Sets the size of the eggs of all species for which this is not
given in the |
max_w |
The largest size of the consumer spectrum. By default this is
set to the largest |
min_w_pp |
The smallest size of the resource spectrum. By default this is set to the smallest value at which any of the consumers can feed. |
pred_kernel |
Optional. An array (species x predator size x prey size) that holds the predation coefficient of each predator at size on each prey size. If not supplied, a default is set as described in section "Setting predation kernel". |
search_vol |
Optional. An array (species x size) holding the search volume for each species at size. If not supplied, a default is set as described in the section "Setting search volume". |
intake_max |
Optional. An array (species x size) holding the maximum intake rate for each species at size. If not supplied, a default is set as described in the section "Setting maximum intake rate". |
metab |
Optional. An array (species x size) holding the metabolic rate for each species at size. If not supplied, a default is set as described in the section "Setting metabolic rate". |
p |
The allometric metabolic exponent. This can be overruled for
individual species by including a |
ext_mort |
Optional. An array (species x size) holding the external mortality rate. If not supplied, a default is set as described in the section "Setting external mortality rate". |
z0pre |
If |
z0exp |
If |
ext_encounter |
Optional. An array (species x size) holding the external
encounter rate. If not supplied, a default is calculated from the |
maturity |
Optional. An array (species x size) that holds the proportion of individuals of each species at size that are mature. If not supplied, a default is set as described in the section "Setting reproduction". |
repro_prop |
Optional. An array (species x size) that holds the proportion of the energy available for growth and reproduction that a mature individual allocates to reproduction for each species at size. If not supplied, a default is set as described in the section "Setting reproduction". |
RDD |
The name of the function calculating the density-dependent
reproduction rate from the density-independent rate. Defaults to
" |
kappa |
The coefficient |
n |
The allometric growth exponent. This can be overruled for individual
species by including a |
resource_rate |
Optional. A vector of per-capita resource birth rate for each size class or a single number giving the coefficient in the power-law for this rate, see "Setting resource dynamics" below. Must be strictly positive. |
resource_capacity |
Optional. Vector of resource intrinsic carrying capacities or coefficient in the power-law for the capacity, see "Setting resource dynamics" below. The resource capacity must not be smaller than the resource abundance. |
lambda |
Used to set power-law exponent for resource capacity if the
|
w_pp_cutoff |
The upper cut off size of the resource spectrum power law
used when |
resource_dynamics |
Optional. Name of the function that determines the resource dynamics by calculating the resource spectrum at the next time step from the current state. |
gear_params |
A data frame with gear-specific parameter values. |
selectivity |
Optional. An array (gear x species x size) that holds the
selectivity of each gear for species and size, |
catchability |
Optional. An array (gear x species) that holds the catchability of
each species by each gear, |
initial_effort |
Optional. A number or a named numeric vector specifying the fishing effort. If a number, the same effort is used for all gears. If a vector, must be named by gear. |
second_order_w |
|
info_level |
Controls the amount of information messages that are shown when the function sets default values for parameters. Higher levels lead to more messages. |
z0 |
|
r_pp |
|
Value
An object of type MizerParams
Species parameters
The only essential argument is a data frame that contains the species parameters. The data frame is arranged species by parameter, so each column of the parameter data frame is a parameter and each row has the values of the parameters for one of the species in the model.
There are two essential columns that must be included in the species
parameter data.frame and that do not have default values: the
species column that should hold strings with the names of the
species and the w_inf column with the von Bertalanffy asymptotic sizes of
the species in grams. (You could alternatively specify the corresponding
length in cm in an l_inf column.) The computational upper size boundary
w_max is not essential; if it is missing it defaults to 1.5 * w_inf. For
backwards compatibility, if w_inf is missing it is taken from the
w_repro_max or w_max column instead.
The species_params dataframe also needs to contain the parameters needed
by any predation kernel function (size selectivity function). This will
be mentioned in the appropriate sections below.
For all other species parameters, mizer will calculate default values if they
are not included in the species parameter data frame. They will be
automatically added when the MizerParams object is created. For these
parameters you can also specify values for only some species and leave the
other entries as NA and the missing values will be set to the defaults.
So the species_params data frame saved in the returned MizerParams object
will differ from the one you supply because it will have the missing
species parameters filled in with default values.
If you are not happy with any of the species parameter values used you can
always change them later with species_params<-().
All the parameters will be mentioned in the following sections.
Setting initial values
The initial values for the species number densities are set using the
function get_initial_n(). These are quite arbitrary and not very close to
the steady state abundances. We intend to improve this in the future.
The initial resource number density N_R(w) is set to a power law with
coefficient kappa (\kappa) and exponent -lambda (-\lambda):
N_R(w) = \kappa\, w^{-\lambda}
for all w less than w_pp_cutoff and zero for sizes at or above
w_pp_cutoff.
Size grid
A size grid is created so that
the log-sizes are equally spaced. The spacing is chosen so that there will be
no_w fish size bins, with the smallest starting at min_w and the largest
starting at max_w. For the resource spectrum there is a larger set of
bins containing additional bins below
min_w, with the same log size. The number of extra bins is such that
min_w_pp comes to lie within the smallest bin.
Units in mizer
Mizer uses grams to measure weight, centimetres to measure lengths, and years to measure time.
Mizer is agnostic about whether abundances are given as
numbers per area,
numbers per volume or
total numbers for the entire study area.
You should make the choice most convenient for your application and then stick with it. If you make choice 1 or 2 you will also have to choose a unit for area or volume. Your choice will then determine the units for some of the parameters. This will be mentioned when the parameters are discussed in the sections below.
Your choice will also affect the units of the quantities you may want to
calculate with the model. For example, the yield will be in grams/year/m^2 in
case 1 if you choose m^2 as your measure of area, in grams/year/m^3 in case 2
if you choose m^3 as your unit of volume, or simply grams/year in case 3. The
same comment applies for other measures, like total biomass, which will be
grams/area in case 1, grams/volume in case 2 or simply grams in case 3. When
mizer puts units on axes in plots, it will choose the units appropriate for
case 3. So for example in plotBiomass() it gives the unit as grams.
You can convert between these choices. For example, if you use case 1, you
need to multiply with the area of the ecosystem to get the total quantity.
If you work with case 2, you need to multiply by both area and the thickness
of the productive layer. In that respect, case 2 is a bit cumbersome. The
function scaleModel() is useful to change the units you are using.
Setting interaction matrix
You do not need to specify an interaction matrix. If you do not, then the predator-prey interactions are purely determined by the size of predator and prey and totally independent of the species of predator and prey.
The interaction matrix \theta_{ij} modifies the interaction of each
pair of species in the model. This can be used for example to allow for
different spatial overlap among the species.
The values in the interaction matrix are used to scale the encountered food
and predation mortality (see on the website the section on predator-prey encounter rate
and on predation mortality).
The first index refers to the predator species and the second to the prey
species.
The interaction matrix is used when calculating the food encounter rate in
getEncounter() and the predation mortality rate in getPredMort(). Its
entries are dimensionless numbers. If all the values in the interaction
matrix are equal then predator-prey interactions are determined entirely by
size-preference.
This function checks that the supplied interaction matrix is valid and then
stores it in the interaction slot of the params object.
The order of the columns and rows of the interaction argument should be
the same as the order in the species params data frame in the params
object. If you supply a named array then the function will check the order
and message if it is different before ignoring the supplied dimnames. If
you supply only column names then these are also used as the row names. One
way of creating your own interaction
matrix is to enter the data using a spreadsheet program and saving it as a
.csv file. The data can then be read into R using the command read.csv().
The interaction of the species with the resource are set via a column
interaction_resource in the species_params data frame. By default this
column is set to all 1s.
Setting predation kernel
Kernel dependent on predator to prey size ratio
If the pred_kernel argument is not supplied, then this function sets a
predation kernel that depends only on the ratio of predator mass to prey
mass, not on the two masses independently. The shape of that kernel is then
determined by the pred_kernel_type column in species_params.
The default for pred_kernel_type is "lognormal". This will call the function
lognormal_pred_kernel() to calculate the predation kernel.
An alternative pred_kernel type is "box", implemented by the function
box_pred_kernel(), and "power_law", implemented by the function
power_law_pred_kernel(). These functions require certain species
parameters in the species_params data frame. For the lognormal kernel these
are beta and sigma, for the box kernel they are ppmr_min
and ppmr_max. They are explained in the help pages for the kernel
functions. Except for beta and sigma, no defaults are set for
these parameters. If they are missing from the species_params data frame then
mizer will issue an error message.
You can use any other string for pred_kernel_type. If for example you
choose "my" then you need to define a function my_pred_kernel that you can
model on the existing functions like lognormal_pred_kernel().
When using a kernel that depends on the predator/prey size ratio only, mizer
does not need to store the entire three dimensional array in the MizerParams
object. Such an array can be very big when there is a large number of size
bins. Instead, mizer only needs to store two two-dimensional arrays that hold
Fourier transforms of the feeding kernel function that allow the encounter
rate and the predation rate to be calculated very efficiently. However, if
you need the full three-dimensional array you can calculate it with the
getPredKernel() function.
Kernel dependent on both predator and prey size
If you want to work with a feeding kernel that depends on predator mass and prey mass independently, you can specify the full feeding kernel as a three-dimensional array (predator species x predator size x prey size).
You should use this option only if a kernel dependent only on the predator/prey mass ratio is not appropriate. Using a kernel dependent on predator/prey mass ratio only allows mizer to use fast Fourier transform methods to significantly reduce the running time of simulations.
The order of the predator species in pred_kernel should be the same
as the order in the species params dataframe in the params object. If you
supply a named array then the function will check the order and warn if it is
different.
Setting search volume
The search volume \gamma_i(w) of an individual of species i
and weight w multiplies the predation kernel when
calculating the encounter rate in getEncounter() and the
predation rate in getPredRate().
The name "search volume" is a bit misleading, because \gamma_i(w) does
not have units of volume. It is simply a parameter that determines the rate
of predation. Its units depend on your choice, see section "Units in mizer".
If you have chosen to work with total abundances, then it is a rate with units
1/year. If you have chosen to work with abundances per m^2 then it has units
of m^2/year. If you have chosen to work with abundances per m^3 then it has
units of m^3/year.
If the search_vol argument is not supplied, then the search volume is
set to
\gamma_i(w) = \gamma_i w^q_i.
The values of \gamma_i (the search volume at 1g) and q_i (the
allometric exponent of the search volume) are taken from the gamma and
q columns in the species parameter dataframe. If the gamma
column is not supplied in the species parameter dataframe, a default is
calculated by the get_gamma_default() function. If the q column is not
supplied, a default of lambda - 2 + n is used. Note that only
for predators of size w = 1 gram is the value of the species parameter
\gamma_i the same as the value of the search volume \gamma_i(w).
If the search_vol slot has a comment and reset = FALSE, then a
recalculation from the species parameters is suppressed and a message is
issued if the recalculated values would differ from the stored ones.
Setting maximum intake rate
The maximum intake rate h_i(w) of an individual of species i and
weight w determines the feeding level, calculated with
getFeedingLevel(). It is measured in grams/year.
If the intake_max argument is not supplied, then the maximum intake
rate is set to
h_i(w) = h_i w^{n_i}.
The values of h_i (the maximum intake rate of an individual of size 1
gram) and n_i (the allometric exponent for the intake rate) are taken
from the h and n columns in the species parameter dataframe. If
the h column is not supplied in the species parameter dataframe, it is
calculated by the get_h_default() function. If the n column is not
supplied, a default of n_i = 3/4 is used.
If h_i is set to Inf, fish of species i will consume all encountered
food.
If the intake_max slot has a comment and reset = FALSE, then a
recalculation from the species parameters is suppressed and a message is
issued if the recalculated values would differ from the stored ones.
Setting metabolic rate
The metabolic rate is subtracted from the energy income rate to calculate
the rate at which energy is available for growth and reproduction, see
getEReproAndGrowth(). It is measured in grams/year.
If the metab argument is not supplied, then for each species the
metabolic rate k(w) for an individual of size w is set to
k(w) = k_s w^p + k w,
where k_s w^p represents the rate of standard metabolism and k w
is the rate at which energy is expended on activity and movement. The values
of k_s, p and k are taken from the ks, p and
k columns in the species parameter dataframe. If any of these
parameters are not supplied, the defaults are k = 0, p = n and
k_s = f_c h \alpha w_{mat}^{n-p},
where f_c is the critical feeding level taken from the fc column
in the species parameter data frame. If the critical feeding level is not
specified, a default of f_c = 0.2 is used.
If the metab slot has a comment and reset = FALSE, then a recalculation
from the species parameters is suppressed and a message is issued if the
recalculated values would differ from the stored ones.
Setting external mortality rate
The external mortality is all the mortality that is not due to fishing or predation by predators included in the model. The external mortality could be due to predation by predators that are not explicitly included in the model (e.g. mammals or seabirds) or due to other causes like illness. It is a rate with units 1/year.
The ext_mort argument allows you to specify an external mortality rate
that depends on species and body size. You can see an example of this in
the Examples section of the help page for setExtMort().
If the ext_mort argument is not supplied, then the external mortality is
taken from the species parameters as
\mu_{ext.i}(w) = z_{0.i} + z_{ext.i} w^{d_i}.
The value of the constant z_0 for each species is taken from the z0
column of the species parameter data frame, if that column exists.
Otherwise it is calculated as
z_{0.i} = {\tt z0pre}_i\, w_{inf}^{\tt z0exp}.
Missing values of z_ext are set to 0 and missing values of d are set to
n - 1.
By default the power law is evaluated at the left bin edges w_j
(point sampling). If the bin_average entry of the second_order_w slot is
TRUE (see second_order_w()), then the z_{ext} w^d term is instead
replaced by its exact average over each bin [w_j, w_{j+1}],
\frac{z_{ext}}{\Delta w_j}\int_{w_j}^{w_{j+1}} w^d\, dw
= z_{ext}\,\frac{w_{j+1}^{d+1} - w_j^{d+1}}{(d+1)\,\Delta w_j},
(with the limiting form z_{ext}\ln(w_{j+1}/w_j)/\Delta w_j when
d = -1). This is the consistent choice in the finite-volume scheme,
where the external mortality multiplies the bin-averaged abundance. The
bin-averaging is applied only to the auto-calculated power-law default; a
user-supplied ext_mort array is left untouched.
Setting external encounter rate
The external encounter rate is the rate at which a predator encounters food that is not explicitly modelled. It is a rate with units mass/year.
The ext_encounter argument allows you to specify an external encounter rate
that depends on species and body size. You can see an example of this in
the Examples section of the help page for setExtEncounter().
If the ext_encounter argument is not supplied, then the external encounter
rate is calculated as a power law:
E_{ext.i}(w) = E_{ext.i}\, w^{n_i}.
The coefficient E_{ext.i} is taken from the E_ext column of the
species parameter data frame, which defaults to 0. The exponent n_i is
taken from the n column of the species parameter data frame.
If the ext_encounter slot has a comment and reset = FALSE, then a
recalculation from the species parameters is suppressed and a message is
issued if the recalculated values would differ from the stored ones.
Setting external diffusion rate
The external diffusion rate allows you to impose additional diffusion beyond the predation-driven diffusion that can be internally modelled by mizer.
The ext_diffusion argument allows you to specify a diffusion rate that
depends on species and body size.
If the ext_diffusion argument is not supplied, then the external diffusion
rate is calculated as a power law:
D_{ext.i}(w) = D_{ext.i}\, w^{n_i+1}.
The coefficient D_{ext.i} is taken from the D_ext column of the
species parameter data frame, which defaults to 0. The exponent
n_i + 1 uses the n column of the species parameter data frame.
If the ext_diffusion slot has a comment and reset = FALSE, then a
recalculation from the species parameters is suppressed and a message is
issued if the recalculated values would differ from the stored ones.
Setting reproduction
For each species and at each size, the proportion \psi of the
available energy
that is invested into reproduction is the product of two factors: the
proportion maturity of individuals that are mature and the proportion
repro_prop of the energy available to a mature individual that is
invested into reproduction. There is a size w_repro_max at which a typical
mature individual invests all of its available energy into reproduction.
This is not a hard ceiling on size: not all individuals are mature at
w_repro_max, and diffusion in the growth process allows some individuals to
grow beyond it, so fish larger than w_repro_max can exist. If you have not
specified the w_repro_max column in the species parameter data frame, then
the von Bertalanffy asymptotic size w_inf is used instead.
Maturity ogive
If the the proportion of individuals that are mature is not supplied via
the maturity argument, then it is set to a sigmoidal
maturity ogive that changes from 0 to 1 at around the maturity size:
{\tt maturity}(w) = \left[1+\left(\frac{w}{w_{mat}}\right)^{-U}\right]^{-1}.
(To avoid clutter, we are not showing the species index in the equations,
although each species has its own maturity ogive.)
The maturity weights are taken from the w_mat column of the
species_params data frame. Any missing maturity weights are set to 1/4 of the
asymptotic size in the w_inf column.
The exponent U determines the steepness of the maturity ogive. By
default it is chosen as U = 10, however this can be overridden by
including a column w_mat25 in the species parameter dataframe that
specifies the weight at which 25% of individuals are mature, which sets
U = \log(3) / \log(w_{mat} / w_{mat25}).
The sigmoidal function given above would strictly reach 0 only
asymptotically and thus have some (negligible) amount of reproduction at
arbitrarily small size.
For computational simplicity, any proportion smaller than
1e-8 is set to 0.
Investment into reproduction
If the the energy available to a mature individual that is
invested into reproduction is not supplied via the repro_prop argument,
it is set to the allometric form
{\tt repro\_prop}(w) =
\min\left(\left(\dfrac{w}{w_{\tt{repro\_max}}}\right)^{m-n},1\right).
Here n is the scaling exponent of the energy income rate. Hence
the exponent m determines the scaling of the investment into
reproduction for mature individuals. By default it is chosen to be
m = 1 so that the rate at which energy is invested into reproduction
scales linearly with the size. This default can be overridden by including a
column m in the species parameter dataframe. The sizes w_{repro\_max}
are taken from the w_repro_max column in the species parameter data frame,
if it exists, or otherwise from the w_inf column.
The total proportion of energy invested into reproduction of an individual
of size w is then
\psi(w) = {\tt maturity}(w){\tt repro\_prop}(w)
In mizer edition 1, at sizes above w_repro_max the value of \psi
is additionally forced to 1, so that all available energy is invested into
reproduction and growth stops. In edition 2 and above this forcing is not
applied, and \psi is determined entirely by the maturity ogive and the
reproductive proportion.
Reproductive efficiency
The reproductive efficiency \epsilon, i.e., the proportion of energy allocated to
reproduction that results in egg biomass, is set through the erepro
column in the species_params data frame. If that is not provided, the default
is set to 1 (which you will want to override). The offspring biomass divided
by the egg biomass gives the rate of egg production, returned by
getRDI():
R_{di} = \frac{\epsilon}{2 w_{min}} \int N(w) E_r(w) \psi(w) \, dw
Density dependence
The stock-recruitment relationship is an emergent phenomenon in mizer, with several sources of density dependence. Firstly, the amount of energy invested into reproduction depends on the energy income of the spawners, which is density-dependent due to competition for prey. Secondly, the proportion of larvae that grow up to recruitment size depends on the larval mortality, which depends on the density of predators, and on larval growth rate, which depends on density of prey.
Finally, to encode all the density dependence in the stock-recruitment
relationship that is not already included in the other two sources of density
dependence, mizer puts the the density-independent rate of egg production
through a density-dependence function. The result is returned by
getRDD(). The name of the density-dependence function is
specified by the RDD argument. The default is the Beverton-Holt
function BevertonHoltRDD(), which requires an R_max column
in the species_params data frame giving the maximum egg production rate. If
this column does not exist, it is initialised to Inf, leading to no
density-dependence. Other functions provided by mizer are
RickerRDD() and SheperdRDD() and you can easily use
these as models for writing your own functions.
Setting fishing
Gears
In mizer, fishing mortality is imposed on species by fishing gears. The
total per-capita fishing mortality (1/year) is obtained by summing over the
mortality from all gears,
\mu_{f.i}(w) = \sum_g F_{g,i}(w),
where the fishing mortality F_{g,i}(w) imposed by gear g on
species i at size w is calculated as:
F_{g,i}(w) = S_{g,i}(w) Q_{g,i} E_{g},
where S is the selectivity by species, gear and size, Q is the
catchability by species and gear and E is the fishing effort by gear.
Selectivity
The selectivity at size of each gear for each species is saved as a three
dimensional array (gear x species x size). Each entry has a range between 0
(that gear is not selecting that species at that size) to 1 (that gear is
selecting all individuals of that species of that size). This three
dimensional array can be specified explicitly via the selectivity
argument, but usually mizer calculates it from the gear_params slot of
the MizerParams object.
To allow the calculation of the selectivity array, the gear_params slot
must be a data frame with one row for each gear-species combination. So if
for example a gear can select three species, then that gear contributes three
rows to the gear_params data frame, one for each species it can select. The
data frame must have columns gear, holding the name of the gear, species,
holding the name of the species, and sel_func, holding the name of the
function that calculates the selectivity curve. Some selectivity functions
are included in the package: knife_edge(), sigmoid_length(),
double_sigmoid_length(), and sigmoid_weight().
Users are able to write their own size-based selectivity function. The first
argument to the function must be w and the function must return a vector of
the selectivity (between 0 and 1) at size.
Each selectivity function may have parameters. Values for these
parameters must be included as columns in the gear parameters data.frame.
The names of the columns must exactly match the names of the corresponding
arguments of the selectivity function. For example, the default selectivity
function is knife_edge() that a has sudden change of selectivity from 0 to 1
at a certain size. In its help page you can see that the knife_edge()
function has arguments w and knife_edge_size. The first argument, w, is
size (the function calculates selectivity at size). All selectivity functions
must have w as the first argument. The values for the other arguments must
be found in the gear parameters data.frame. So for the knife_edge()
function there should be a knife_edge_size column. Because knife_edge()
is the default selectivity function, the knife_edge_size argument has a
default value = w_mat.
The most commonly-used selectivity function is sigmoid_length(). It has a
smooth transition from 0 to 1 at a certain size. The sigmoid_length()
function has the two parameters l50 and l25 that are the lengths in cm at
which 50% or 25% of the fish are selected by the gear. If you choose this
selectivity function then the l50 and l25 columns must be included in the
gear parameters data.frame.
In case each species is only selected by one gear, the columns of the
gear_params data frame can alternatively be provided as columns of the
species_params data frame, if this is more convenient for the user to set
up. Mizer will then copy these columns over to create the gear_params data
frame when it creates the MizerParams object. However changing these columns
in the species parameter data frame later will not update the gear_params
data frame.
Catchability
Catchability is used as an additional factor to make the link between gear selectivity, fishing effort and fishing mortality. For example, it can be set so that an effort of 1 gives a desired fishing mortality. In this way effort can then be specified relative to a 'base effort', e.g. the effort in a particular year.
Catchability is stored as a two dimensional array (gear x species). This can
either be provided explicitly via the catchability argument, or the
information can be provided via a catchability column in the gear_params
data frame.
In the case where each species is selected by only a single gear, the
catchability column can also be provided in the species_params data
frame. Mizer will then copy this over to the gear_params data frame when
the MizerParams object is created.
Effort
The initial fishing effort is stored in the MizerParams object. If it is
not supplied, it is set to zero. The initial effort can be overruled when
the simulation is run with project(), where it is also possible to specify
an effort that varies through time.
Setting resource dynamics
The resource_dynamics argument allows you to choose the resource dynamics
function. By default, mizer uses a semichemostat model to describe the
resource dynamics in each size class independently. This semichemostat
dynamics is implemented by the function resource_semichemostat(). You can
change that to use a logistic model implemented by resource_logistic() or
you can use resource_constant() which keeps the resource constant or you
can write your own function.
Both the resource_semichemostat() and the resource_logistic() dynamics
are parametrised in terms of a size-dependent birth rate r_R(w) and a
size-dependent capacity c_R. The help pages of these functions give
the details.
The resource_rate argument can be a vector (with the same length as
w_full(params)) specifying the intrinsic resource birth rate for each size
class. Alternatively it can be a single number that is used as the
coefficient in a power law: then the intrinsic birth rate r_R(w) at
size w is set to
r_R(w) = r_R w^{n-1}.
The power-law exponent n is taken from the n argument.
The resource_capacity argument can be a vector specifying the intrinsic
resource carrying capacity for each size class. Alternatively it can be a
single number that is used as the coefficient in a truncated power
law: then the intrinsic carrying capacity c_R(w) at size w
is set to
c_R(w) = c_R\, w^{-\lambda}
for all w less than w_pp_cutoff and zero for larger sizes.
The power-law exponent \lambda is taken from the lambda argument.
The values for lambda, n and w_pp_cutoff are stored in a list
in the resource_params slot of the MizerParams object so that they can be
re-used automatically in the future. If you specify resource_rate or
resource_capacity as a single number, that coefficient is likewise stored,
as r_pp and kappa respectively. That list can be accessed with
resource_params().
See Also
Other functions for setting up models:
newCommunityParams(),
newSingleSpeciesParams(),
newTraitParams()
Examples
params <- newMultispeciesParams(NS_species_params)
Set up parameters for a single species in a power-law background
Description
This functions creates a MizerParams object with a single
species. This species is embedded in a fixed power-law community spectrum
N_c(w) = \kappa w^{-\lambda}
This community provides the food income for the species. Cannibalism is switched off. The predation mortality arises only from the predators in the power-law community and it is assumed that the predators in the community have the same feeding parameters as the foreground species. The function has many arguments, all of which have default values.
Usage
newSingleSpeciesParams(
species_name = "Target species",
w_max = 100,
w_min = 0.001,
eta = 10^(-0.6),
w_mat = w_max * eta,
no_w = log10(w_max/w_min) * 20 + 1,
n = 3/4,
p = n,
lambda = 2.05,
kappa = 0.005,
alpha = 0.4,
h = 30,
beta = 100,
sigma = 1.3,
f0 = 0.6,
fc = 0.25,
ks = NA,
gamma = NA,
ext_mort_prop = 0,
reproduction_level = 0,
second_order_w = FALSE,
R_factor = deprecated(),
w_inf = deprecated(),
k_vb = deprecated()
)
Arguments
species_name |
A string with a name for the species. Will be used in plot legends. |
w_max |
Maximum size of species |
w_min |
Egg size of species |
eta |
Ratio between maturity size |
w_mat |
Maturity size of species. Default value is
|
no_w |
The number of size bins in the community spectrum. These bins will be equally spaced on a logarithmic scale. Default value is such that there are 20 bins for each factor of 10 in weight. |
n |
Scaling exponent of the maximum intake rate. |
p |
Scaling exponent of the standard metabolic rate. By default this is
equal to the exponent |
lambda |
Exponent of the abundance power law. |
kappa |
Coefficient in abundance power law. |
alpha |
The assimilation efficiency. |
h |
Maximum food intake rate. |
beta |
Preferred predator prey mass ratio. |
sigma |
Width of prey size preference. |
f0 |
Expected average feeding level. Used to set |
fc |
Critical feeding level. Used to determine |
ks |
Standard metabolism coefficient. If not provided, default will be
calculated from critical feeding level argument |
gamma |
Volumetric search rate. If not provided, default is determined
by |
ext_mort_prop |
The proportion of the total mortality that comes from external mortality, i.e., from sources not explicitly modelled. A number in the interval [0, 1). |
reproduction_level |
A number between 0 and 1 that determines the
level of density dependence in reproduction, see |
second_order_w |
|
R_factor |
|
w_inf |
|
k_vb |
|
Details
In addition to setting up the parameters, this function also sets up an initial condition that is close to steady state, under the assumption of no fishing.
The function rounds no_w to the nearest integer and increases it if
necessary so that there are at least 5 size bins per factor 10 in body
size. It requires w_min < w_mat < w_max, ext_mort_prop in [0, 1),
positive values for n, lambda, kappa, alpha, h, beta, sigma
and f0, and fc between 0 and f0 if fc is supplied. If gamma is
supplied then f0 is ignored. The function stops if the resulting feeding
level is not sufficient to maintain the species.
The returned model has a single foreground species with cannibalism switched
off and a fixed power-law background community that provides both food and
predation mortality. The initial species spectrum is scaled so that its
maximum abundance is half the background abundance at the corresponding
size, and erepro is then adjusted so the initial state satisfies the egg
boundary condition.
The diffusion rate is set to 0. Because growth is therefore deterministic,
no individual grows beyond w_repro_max, the size at which all available
energy is invested into reproduction. The upper boundary of the size grid is
therefore placed at that size, so that w_max = w_repro_max, instead of the
1.5 * w_repro_max headroom that newMultispeciesParams() leaves to
accommodate the stochastic growth produced by diffusion. This choice will be
revisited once these constructors gain a diffusion parameter, see
https://github.com/sizespectrum/mizer/issues/339.
Value
An object of type MizerParams
See Also
Other functions for setting up models:
newCommunityParams(),
newMultispeciesParams(),
newTraitParams()
Examples
params <- newSingleSpeciesParams()
sim <- project(params, t_max = 5, effort = 0)
plotSpectra(sim)
Set up parameters for a trait-based multispecies model
Description
This functions creates a MizerParams object describing a trait-based
model. This is a simplification of the general size-based model used in
mizer in which the species-specific parameters are the same for all
species, except for the maximum size, which is considered the most
important trait characterizing a species. Other parameters are related to the
maximum size. For example, the size at maturity is given by w_max *
eta, where eta is the same for all species. For the trait-based model
the number of species is not important. For applications of the trait-based
model see Andersen & Pedersen (2010). See the mizer website for more
details and examples of the trait-based model.
Usage
newTraitParams(
no_sp = 11,
min_w_max = 10,
max_w_max = 10^4,
min_w = 10^(-3),
max_w = max_w_max,
eta = 10^(-0.6),
min_w_mat = min_w_max * eta,
no_w = round(log10(max_w_max/min_w) * 20 + 1),
min_w_pp = 1e-10,
w_pp_cutoff = min_w_mat,
n = 2/3,
p = n,
lambda = 2.05,
r_pp = 0.1,
kappa = 0.005,
alpha = 0.4,
h = 40,
beta = 100,
sigma = 1.3,
f0 = 0.6,
fc = 0.25,
ks = NA,
gamma = NA,
ext_mort_prop = 0,
reproduction_level = 1/4,
R_factor = deprecated(),
gear_names = "knife_edge_gear",
knife_edge_size = 1000,
egg_size_scaling = FALSE,
resource_scaling = FALSE,
perfect_scaling = FALSE,
second_order_w = FALSE,
min_w_inf = deprecated(),
max_w_inf = deprecated(),
info_level = 2
)
Arguments
no_sp |
The number of species in the model. |
min_w_max |
The maximum size of the smallest species in the community. This will be rounded to lie on a grid point. |
max_w_max |
The maximum size of the largest species in the community. This will be rounded to lie on a grid point. |
min_w |
The size of the the egg of the smallest species. This also defines the start of the community size spectrum. |
max_w |
The largest size in the model. By default this is set to the
largest maximum size |
eta |
Ratio between maturity size and maximum size of a species.
Ignored if |
min_w_mat |
The maturity size of the smallest species. Default value is
|
no_w |
The number of size bins in the community spectrum. These bins will be equally spaced on a logarithmic scale. Default value is such that there are 20 bins for each factor of 10 in weight. |
min_w_pp |
The smallest size of the resource spectrum. By default this is set to the smallest value at which any of the consumers can feed. |
w_pp_cutoff |
The cutoff used when truncating the constructed resource
spectrum. Resource abundance is retained only up to the largest grid point
below this value. If |
n |
Scaling exponent of the maximum intake rate. |
p |
Scaling exponent of the standard metabolic rate. By default this is
equal to the exponent |
lambda |
Exponent of the abundance power law. |
r_pp |
Growth rate parameter for the resource spectrum. |
kappa |
Coefficient in abundance power law. |
alpha |
The assimilation efficiency. |
h |
Maximum food intake rate. |
beta |
Preferred predator prey mass ratio. |
sigma |
Width of prey size preference. |
f0 |
Expected average feeding level. Used to set |
fc |
Critical feeding level. Used to determine |
ks |
Standard metabolism coefficient. If not provided, default will be
calculated from critical feeding level argument |
gamma |
Volumetric search rate. If not provided, default is determined
by |
ext_mort_prop |
The proportion of the total mortality that comes from external mortality, i.e., from sources not explicitly modelled. A number in the interval [0, 1). |
reproduction_level |
A number between 0 and 1 that determines the
level of density dependence in reproduction, see |
R_factor |
|
gear_names |
The names of the fishing gears for each species. Either a
single name used for all species or a character vector of length |
knife_edge_size |
The minimum size at which the gear or gears select
fish. Either a single value used for all species or a vector of length
|
egg_size_scaling |
|
resource_scaling |
|
perfect_scaling |
|
second_order_w |
|
min_w_inf |
|
max_w_inf |
|
info_level |
Controls the amount of information messages that are shown. Higher levels lead to more messages. |
Details
The function has many arguments, all of which have default values. Of particular interest to the user are the number of species in the model and the minimum and maximum sizes.
The characteristic weights of the smallest species are defined by
min_w (egg size), min_w_mat (maturity size) and
min_w_max (maximum size). The maximum sizes of
the no_sp species
are logarithmically evenly spaced, ranging from min_w_max to
max_w_max.
Similarly the maturity sizes of the species are logarithmically evenly
spaced, so that the ratio eta between maturity size and maximum
size is the same for all species. If egg_size_scaling = TRUE then also
the ratio between maximum size and egg size is the same for all species.
Otherwise all species have the same egg size.
In addition to setting up the parameters, this function also sets up an initial condition that is close to steady state.
The search rate coefficient gamma is calculated using the expected
feeding level, f0.
The diffusion rate is set to 0. Because growth is therefore deterministic,
no individual grows beyond w_repro_max, the size at which all available
energy is invested into reproduction. The upper boundary of the size grid is
therefore placed at that size, so that w_max = w_repro_max, instead of the
1.5 * w_repro_max headroom that newMultispeciesParams() leaves to
accommodate the stochastic growth produced by diffusion. This choice will be
revisited once these constructors gain a diffusion parameter, see
https://github.com/sizespectrum/mizer/issues/339.
The option of including fishing is given, but the steady state may loose its
natural stability if too much fishing is included. In such a case the user
may wish to include stabilising effects (like reproduction_level) to ensure
the steady state is stable. Fishing selectivity is modelled as a knife-edge
function with one parameter, knife_edge_size, which is the size at which
species are selected. Each species can either be fished by the same gear
(knife_edge_size has a length of 1) or by a different gear (the length of
knife_edge_size has the same length as the number of species and the order
of selectivity size is that of the maximum size).
The resulting MizerParams object can be projected forward using
project() like any other MizerParams object. When projecting
the model it may be necessary to reduce dt below 0.1 to avoid any
instabilities with the solver. You can check this by plotting the biomass or
abundance through time after the projection.
Value
An object of type MizerParams
See Also
Other functions for setting up models:
newCommunityParams(),
newMultispeciesParams(),
newSingleSpeciesParams()
Examples
params <- newTraitParams()
sim <- project(params, t_max = 5, effort = 0)
plotSpectra(sim)
Give density-independent reproduction rate
Description
Simply returns its rdi argument.
Usage
noRDD(rdi, ...)
Arguments
rdi |
Vector of density-independent reproduction rates
|
... |
Not used. |
Value
Vector of density-dependent reproduction rates.
See Also
Other functions calculating density-dependent reproduction rate:
BevertonHoltRDD(),
RickerRDD(),
SheperdRDD(),
constantEggRDI(),
constantRDD()
Get the extension chain stored in a mizer object
Description
Get the extension chain stored in a mizer object
Usage
objectExtensions(object)
Arguments
object |
A |
Value
A named character vector of extensions, or an empty character vector if the object carries no extensions.
Parse the log-axis arguments of a mizer plot function
Description
Internal helper that resolves the various ways of specifying which axes
should use a logarithmic scale into a consistent pair of logical flags. It
is exported so that extension packages (such as mizerMR) can reuse it in
their own array plot() methods.
Usage
parsePlotLog(log, log_x = FALSE, log_y = FALSE)
Arguments
log |
Either |
log_x, log_y |
Default logical flags used when |
Value
A list with logical components log_x and log_y.
Parse the log argument for time-series plots
Description
Translates the log argument of the time-series plotting functions into a
list of logical flags for the x and y axes, falling back to log_x and
log_y when log is NULL. For backwards compatibility a single logical
log value sets only log_y.
Usage
parseTimePlotLog(log, log_x, log_y)
Arguments
log |
|
log_x, log_y |
Default logical flags used when |
Value
A list with logical elements log_x and log_y.
Plot mizer arrays
Description
Many mizer functions return values that depend on species and either size or
time. plot() creates a ggplot2 figure with one line for each species
showing the values against size or against time (depending on the type of
output). plotHover() creates an interactive version of the same figure.
Details
This works because the mizer functions that give values that depend on
species and size return an ArraySpeciesBySize object and those that
give values that depend on species and time return an ArrayTimeBySpecies
object. These objects have attributes that store the name of the value,
its units, and a reference to the MizerParams object that the value was
computed from. This allows the plots to be automatically labelled and
coloured appropriately.
To compare two mizer arrays in a single plot, use plot2(). To show the
relative difference between two arrays, use plotRelative().
All methods return a ggplot2 object, unless return_data = TRUE, in
which case they return the underlying data frame instead. plotHover()
returns a plotly object.
Arguments used by all methods:
speciesCharacter vector of species to include.
NULL(default) means all species.highlightName or vector of names of the species to be highlighted.
totalA boolean value that determines whether the total over all selected species is plotted as well. Default is
FALSE.backgroundA boolean value that determines whether background species are included. Ignored if the model does not contain background species. Default is
TRUE.return_dataIf
TRUE, return the data frame instead of the plot.log_xIf
TRUE, use a log10 x-axis. The default depends on the method; see its own help page.log_yIf
TRUE, use a log10 y-axis. The default depends on the method; see its own help page.logCharacter string specifying which axes should use log10 scales, in the same form as the base
plot()argument. For example,"x","y","xy"or"". If supplied, this overrideslog_xandlog_y.ylimA numeric vector of length two providing lower and upper limits for the value (y) axis. Use
NAto refer to the existing minimum or maximum.y_ticksThe approximate number of ticks desired on the y axis.
Additional arguments for plot.ArraySpeciesBySize() and
plot.ArrayTimeBySpeciesBySize():
all.sizesIf
FALSE(default), values outside a species' size range (w_mintow_max) are removed.wlimA numeric vector of length two providing lower and upper limits for the weight (x) axis. Use
NAto refer to the existing minimum or maximum.llimA numeric vector of length two providing lower and upper limits for the length (x) axis when
size_axis = "l". UseNAto refer to the existing minimum or maximum.size_axisWhether to plot size as weight (
"w", default) or length ("l"), using the allometric weight-length relationship.
Additional argument for plot.ArrayTimeBySpecies():
tlimA numeric vector of length two providing lower and upper limits for the time axis, e.g.
c(1980, 2000). UseNAto apply no limit at that end. Default isc(NA, NA).
Additional argument for plot.ArrayTimeBySpeciesBySize() and
plot.ArrayTimeByResourceBySize():
timeThe time to display. Default (
NULL) is the final time step.
See the individual method help pages for each method's exact arguments and
defaults: plot.ArraySpeciesBySize(), plot.ArrayTimeBySpecies(),
plot.ArrayTimeBySpeciesBySize(), plot.ArrayResourceBySize(),
plot.ArrayTimeByResourceBySize().
See Also
Other plotting functions:
addPlot(),
animate(),
plot2(),
plotBiomass(),
plotCDF(),
plotCDF2(),
plotDiet(),
plotFMort(),
plotFeedingLevel(),
plotGrowthCurves(),
plotMizerParams,
plotMizerSim,
plotPredMort(),
plotRelative(),
plotSpectra(),
plotSpectra2(),
plotSpectraRelative(),
plotYield(),
plotYieldGear(),
plotting_functions
Examples
plot(getEncounter(NS_params))
plot(getFeedingLevel(NS_params), species = c("Cod", "Herring"))
plot(getPredMort(NS_params), species = c("Cod", "Herring"),
size_axis = "l")
plot(getBiomass(NS_sim))
plot(getBiomass(NS_sim), species = c("Cod", "Herring"), total = TRUE)
plot(getYield(NS_sim), species = c("Cod", "Herring"))
plot(getFMort(NS_sim), time = 2010)
plot(getResourceMort(NS_params))
plot(initialNResource(NS_params))
plot(NResource(NS_sim))
Plot method for ArrayResourceBySize objects
Description
See plot() for an overview of the mizer plotting system and the
arguments shared by all of its methods.
Usage
## S3 method for class 'ArrayResourceBySize'
plot(
x,
return_data = FALSE,
log_x = TRUE,
log_y = TRUE,
log = NULL,
wlim = c(NA, NA),
ylim = c(NA, NA),
y_ticks = 6,
...
)
Arguments
x |
An |
return_data |
If |
log_x |
If |
log_y |
If |
log |
Character string specifying which axes should use log10
scales, in the same form as the base |
wlim |
A numeric vector of length two providing lower and upper
limits for the weight (x) axis. Use |
ylim |
A numeric vector of length two providing lower and upper
limits for the value (y) axis. Use |
y_ticks |
The approximate number of ticks desired on the y axis. |
... |
Unused. |
Value
A ggplot2 object, unless return_data = TRUE, in which case a
data frame is returned.
Examples
plot(getResourceMort(NS_params))
plot(initialNResource(NS_params))
Plot method for ArraySpeciesBySize objects
Description
See plot() for an overview of the mizer plotting system and the
arguments shared by all of its methods.
Usage
## S3 method for class 'ArraySpeciesBySize'
plot(
x,
species = NULL,
all.sizes = FALSE,
highlight = NULL,
return_data = FALSE,
log_x = TRUE,
log_y = FALSE,
log = NULL,
wlim = c(NA, NA),
llim = c(NA, NA),
ylim = c(NA, NA),
size_axis = c("w", "l"),
total = FALSE,
background = TRUE,
y_ticks = 6,
...
)
Arguments
x |
An |
species |
Character vector of species to include. |
all.sizes |
If |
highlight |
Name or vector of names of the species to be highlighted. |
return_data |
If |
log_x |
If |
log_y |
If |
log |
Character string specifying which axes should use log10
scales, in the same form as the base |
wlim |
A numeric vector of length two providing lower and upper
limits for the weight (x) axis. Use |
llim |
A numeric vector of length two providing lower and upper
limits for the length (x) axis when |
ylim |
A numeric vector of length two providing lower and upper
limits for the value (y) axis. Use |
size_axis |
Whether to plot size as weight ( |
total |
A boolean value that determines whether the total over
all selected species is plotted as well. Default is |
background |
A boolean value that determines whether background
species are included. Ignored if the model does not contain background
species. Default is |
y_ticks |
The approximate number of ticks desired on the y axis. |
... |
Unused. |
Value
A ggplot2 object, unless return_data = TRUE, in which case a
data frame is returned.
Examples
plot(getEncounter(NS_params))
plot(getFeedingLevel(NS_params), species = c("Cod", "Herring"))
plot(getPredMort(NS_params), species = c("Cod", "Herring"),
size_axis = "l")
Plot method for ArrayTimeByResourceBySize objects
Description
See plot() for an overview of the mizer plotting system. This method
plots a single time slice, by first extracting it as an
ArrayResourceBySize object and delegating to
plot.ArrayResourceBySize(), which the further arguments in ... are
passed on to.
Usage
## S3 method for class 'ArrayTimeByResourceBySize'
plot(x, time = NULL, ...)
Arguments
x |
An |
time |
The time to display. Default ( |
... |
Passed on to |
Value
A ggplot2 object, unless return_data = TRUE, in which case a
data frame is returned.
Examples
plot(NResource(NS_sim))
Plot method for ArrayTimeBySpecies objects
Description
See plot() for an overview of the mizer plotting system and the
arguments shared by all of its methods.
Usage
## S3 method for class 'ArrayTimeBySpecies'
plot(
x,
species = NULL,
tlim = c(NA, NA),
y_ticks = 6,
ylim = c(NA, NA),
total = FALSE,
background = TRUE,
highlight = NULL,
log_x = FALSE,
log_y = TRUE,
log = NULL,
return_data = FALSE,
...
)
Arguments
x |
An |
species |
Character vector of species to include. |
tlim |
A numeric vector of length two providing lower and upper
limits for the time axis, e.g. |
y_ticks |
The approximate number of ticks desired on the y axis. |
ylim |
A numeric vector of length two providing lower and upper
limits for the value (y) axis. Use |
total |
A boolean value that determines whether the total over
all selected species is plotted as well. Default is |
background |
A boolean value that determines whether background
species are included. Ignored if the model does not contain background
species. Default is |
highlight |
Name or vector of names of the species to be highlighted. |
log_x |
If |
log_y |
If |
log |
Character string specifying which axes should use log10
scales, in the same form as the base |
return_data |
If |
... |
Unused. |
Value
A ggplot2 object, unless return_data = TRUE, in which case a
data frame is returned.
Examples
plot(getBiomass(NS_sim))
plot(getBiomass(NS_sim), species = c("Cod", "Herring"), total = TRUE)
plot(getYield(NS_sim), species = c("Cod", "Herring"))
Plot method for ArrayTimeBySpeciesBySize objects
Description
See plot() for an overview of the mizer plotting system and the
arguments shared by all of its methods. This method plots a single time
slice, by first extracting it as an ArraySpeciesBySize object and
delegating to plot.ArraySpeciesBySize().
Usage
## S3 method for class 'ArrayTimeBySpeciesBySize'
plot(
x,
species = NULL,
time = NULL,
all.sizes = FALSE,
highlight = NULL,
return_data = FALSE,
log_x = TRUE,
log_y = FALSE,
log = NULL,
wlim = c(NA, NA),
llim = c(NA, NA),
ylim = c(NA, NA),
size_axis = c("w", "l"),
total = FALSE,
background = TRUE,
y_ticks = 6,
...
)
Arguments
x |
An |
species |
Character vector of species to include. |
time |
The time to display. Default ( |
all.sizes |
If |
highlight |
Name or vector of names of the species to be highlighted. |
return_data |
If |
log_x |
If |
log_y |
If |
log |
Character string specifying which axes should use log10
scales, in the same form as the base |
wlim |
A numeric vector of length two providing lower and upper
limits for the weight (x) axis. Use |
llim |
A numeric vector of length two providing lower and upper
limits for the length (x) axis when |
ylim |
A numeric vector of length two providing lower and upper
limits for the value (y) axis. Use |
size_axis |
Whether to plot size as weight ( |
total |
A boolean value that determines whether the total over
all selected species is plotted as well. Default is |
background |
A boolean value that determines whether background
species are included. Ignored if the model does not contain background
species. Default is |
y_ticks |
The approximate number of ticks desired on the y axis. |
... |
Unused. |
Value
A ggplot2 object, unless return_data = TRUE, in which case a
data frame is returned.
Examples
plot(getFMort(NS_sim), time = 2010)
Compare two mizer arrays in a single plot
Description
plot2() compares two compatible mizer array objects in a single ggplot.
Colours identify species or groups, and linetype identifies which object
the values came from.
Usage
plot2(
x,
y,
name1 = "First",
name2 = "Second",
species = NULL,
log_x,
log_y,
log = NULL,
ylim = c(NA, NA),
total = FALSE,
background = TRUE,
y_ticks = 6,
...
)
Arguments
x |
The first of two compatible mizer array objects to compare.
Can be an |
y |
The second mizer array object, compatible with |
name1, name2 |
Labels for the two objects, used in the linetype legend. |
species |
Character vector of species to include. |
log_x |
If |
log_y |
If |
log |
Character string specifying which axes should use log10 scales,
in the same form as the base |
ylim |
A numeric vector of length two providing lower and upper limits
for the value (y) axis. Use |
total |
A boolean value that determines whether the total over all
selected species is plotted as well. Default is |
background |
A boolean value that determines whether background species
are included. Ignored if the model does not contain background species.
Default is |
y_ticks |
The approximate number of ticks desired on the y axis. |
... |
Further arguments used by only some of the methods: For
For
For
|
Value
A ggplot2 object.
See Also
Other plotting functions:
addPlot(),
animate(),
plot,
plotBiomass(),
plotCDF(),
plotCDF2(),
plotDiet(),
plotFMort(),
plotFeedingLevel(),
plotGrowthCurves(),
plotMizerParams,
plotMizerSim,
plotPredMort(),
plotRelative(),
plotSpectra(),
plotSpectra2(),
plotSpectraRelative(),
plotYield(),
plotYieldGear(),
plotting_functions
Examples
plot2(getEncounter(NS_params), getEncounter(NS_params))
Plot the biomass of species through time
Description
After running a projection, the biomass of each species can be plotted
against time. The biomass is calculated within user defined size limits
(see getBiomass()).
Usage
plotBiomass(
object,
species = NULL,
tlim = c(NA, NA),
y_ticks = 6,
ylim = c(NA, NA),
total = FALSE,
background = TRUE,
highlight = NULL,
log = NULL,
return_data = FALSE,
log_x = FALSE,
log_y = TRUE,
use_cutoff = FALSE,
...
)
Arguments
object |
An object of class MizerSim |
species |
The species to be selected. Optional. By default all target species are selected. A vector of species names, or a numeric vector with the species indices, or a logical vector indicating for each species whether it is to be selected (TRUE) or not. |
tlim |
A numeric vector of length two providing lower and upper limits
for the time axis, e.g. |
y_ticks |
The approximate number of ticks desired on the y axis. |
ylim |
A numeric vector of length two providing lower and upper limits
for the y axis. Use |
total |
A boolean value that determines whether the total biomass from all species is plotted as well. Default is FALSE. |
background |
A boolean value that determines whether background species are included. Ignored if the model does not contain background species. Default is TRUE. |
highlight |
Name or vector of names of the species to be highlighted. |
log |
Character string specifying which axes should use log10 scales,
in the same form as the base |
return_data |
A boolean value that determines whether the formatted data used for the plot is returned instead of the plot itself. Default is FALSE. |
log_x |
If |
log_y |
If |
use_cutoff |
If TRUE, the |
... |
Arguments setting the size range over which the biomass is
calculated (see
|
Value
A ggplot2 object, unless return_data = TRUE, in which case a data
frame with the four variables 'Year', 'Biomass', 'Species', 'Legend' is
returned.
See Also
plotting_functions, getBiomass()
Other plotting functions:
addPlot(),
animate(),
plot,
plot2(),
plotCDF(),
plotCDF2(),
plotDiet(),
plotFMort(),
plotFeedingLevel(),
plotGrowthCurves(),
plotMizerParams,
plotMizerSim,
plotPredMort(),
plotRelative(),
plotSpectra(),
plotSpectra2(),
plotSpectraRelative(),
plotYield(),
plotYieldGear(),
plotting_functions
Examples
plotBiomass(NS_sim)
plotBiomass(NS_sim, species = c("Sandeel", "Herring"), total = TRUE)
plotBiomass(NS_sim, tlim = c(1980, 1990))
# Returning the data frame
fr <- plotBiomass(NS_sim, return_data = TRUE)
str(fr)
Plotting observed vs. model biomass data
Description
If biomass observations are available for at least some species via the
biomass_observed column in the species parameter data frame, this function
plots the biomass of each species in the model against the observed
biomasses. When called with a MizerSim object, the plot will use the model
biomasses predicted for the final time step in the simulation. ratio
defaults to FALSE.
Usage
plotBiomassObservedVsModel(
object,
species = NULL,
ratio = FALSE,
log_scale = TRUE,
return_data = FALSE,
labels = TRUE,
show_unobserved = FALSE,
...
)
Arguments
object |
An object of class MizerParams or MizerSim. |
species |
The species to be included. Optional. By default all observed biomasses will be included. A vector of species names, or a numeric vector with the species indices, or a logical vector indicating for each species whether it is to be included (TRUE) or not. |
ratio |
Whether to plot model biomass vs. observed biomass (FALSE) or the ratio of model : observed biomass (TRUE). Default is FALSE. |
log_scale |
Whether to plot on the log10 scale (TRUE) or not (FALSE). For the non-ratio plot this applies for both axes, for the ratio plot only the x-axis is on the log10 scale. Default is TRUE. |
return_data |
Whether to return the data frame for the plot (TRUE) or not (FALSE). Default is FALSE. |
labels |
Whether to show text labels for each species (TRUE) or not (FALSE). Default is TRUE. |
show_unobserved |
Whether to include also species for which no biomass observation is available. If TRUE, these species will be shown as if their observed biomass was equal to the model biomass. |
... |
For |
Details
Before you can use this function you will need to have added a
biomass_observed column to your model which gives the observed biomass in
grams. For species for which you have no observed biomass, you should set
the value in the biomass_observed column to 0 or NA.
Biomass observations usually only include individuals above a certain size.
This size should be specified in a biomass_cutoff column of the species
parameter data frame. If this is missing, it is assumed that all sizes are
included in the observed biomass, i.e., it includes larval biomass.
The total relative error is shown in the caption of the plot, calculated by
TRE = \sum_i|1-\rm{ratio_i}|
where
\rm{ratio_i} is the ratio of model biomass / observed
biomass for species i.
Value
A ggplot2 object with the plot of model biomass by species compared
to observed biomass. If return_data = TRUE, the data frame used to
create the plot is returned instead of the plot.
Examples
# create an example
params <- NS_params
species_params(params)$biomass_observed <-
c(0.8, 61, 12, 35, 1.6, NA, 10, 7.6, 135, 60, 30, NA)
species_params(params)$biomass_cutoff <- 10
params <- calibrateBiomass(params)
# Plot with default options
plotBiomassObservedVsModel(params, ratio = FALSE)
# Plot including also species without observations
plotBiomassObservedVsModel(params, show_unobserved = TRUE, ratio = FALSE)
# Show the ratio instead
plotBiomassObservedVsModel(params, ratio = TRUE)
Plot cumulative abundance or biomass distributions
Description
plotCDF() plots the cumulative distribution over body size from small to
large sizes. It uses the same spectra data preparation as plotSpectra().
The density is first multiplied by w^power, then integrated over size.
With normalise = TRUE, each curve is divided by its final value so that it
ends at 1.
Usage
plotCDF(
object,
species = NULL,
wlim = c(NA, NA),
llim = c(NA, NA),
ylim = c(NA, NA),
power = 1,
biomass = TRUE,
total = FALSE,
resource = FALSE,
background = TRUE,
highlight = NULL,
normalise = TRUE,
log_x = TRUE,
log_y = FALSE,
log = NULL,
size_axis = c("w", "l"),
return_data = FALSE,
...
)
Arguments
object |
An object of class MizerSim or MizerParams. |
species |
The species to be selected. Optional. By default all target species are selected. A vector of species names, or a numeric vector with the species indices, or a logical vector indicating for each species whether it is to be selected (TRUE) or not. |
wlim |
A numeric vector of length two providing lower and upper limits
for the w axis. Use NA for the default: the lower default is
|
llim |
A numeric vector of length two providing lower and upper limits
for the length axis when |
ylim |
A numeric vector of length two providing lower and upper limits
for the y axis. Use NA to auto-scale to the data range. Values below 1e-20
are always filtered out from the data regardless of |
power |
The abundance is plotted as the number density times the weight
raised to |
biomass |
|
total |
A boolean value that determines whether the total over all species in the system is plotted as well. Note that even if the plot only shows a selection of species, the total is including all species. Default is FALSE. |
resource |
A boolean value that determines whether resource is included. Default is FALSE. |
background |
A boolean value that determines whether background species are included. Ignored if the model does not contain background species. Default is TRUE. |
highlight |
Name or vector of names of the species to be highlighted by being plotted with thicker lines. |
normalise |
If |
log_x |
If |
log_y |
If |
log |
Character string specifying which axes should use a log10 scale,
in the same form as the base |
size_axis |
Whether to plot size as weight ( |
return_data |
A boolean value that determines whether the formatted data used for the plot is returned instead of the plot itself. Default is FALSE. |
... |
Further arguments used by only some of the methods: For
|
Details
plotlyCDF() is the interactive plotly version. To compare cumulative
distributions from two objects, use plotCDF2().
Value
A ggplot2 object, unless return_data = TRUE, in which case a data
frame with the four variables 'w' (or 'l' if size_axis = "l"), 'value',
'Species', 'Legend' is returned. plotlyCDF() returns a plotly object.
See Also
Other plotting functions:
addPlot(),
animate(),
plot,
plot2(),
plotBiomass(),
plotCDF2(),
plotDiet(),
plotFMort(),
plotFeedingLevel(),
plotGrowthCurves(),
plotMizerParams,
plotMizerSim,
plotPredMort(),
plotRelative(),
plotSpectra(),
plotSpectra2(),
plotSpectraRelative(),
plotYield(),
plotYieldGear(),
plotting_functions
Examples
plotCDF(NS_params, species = c("Cod", "Herring"))
plotCDF(NS_sim, power = 0, normalise = FALSE)
Compare cumulative abundance or biomass distributions from two objects
Description
plotCDF2() compares cumulative distributions from two MizerParams or
MizerSim objects in a single plot. Colours identify species or groups and
linetype identifies the object.
Usage
plotCDF2(
object1,
object2,
name1 = "First",
name2 = "Second",
species = NULL,
wlim = c(NA, NA),
llim = c(NA, NA),
ylim = c(NA, NA),
power = 1,
biomass = TRUE,
total = FALSE,
resource = FALSE,
background = TRUE,
highlight = NULL,
normalise = TRUE,
log_x = TRUE,
log_y = FALSE,
log = NULL,
size_axis = c("w", "l"),
...
)
Arguments
object1 |
First |
object2 |
Second |
name1, name2 |
Labels for the two objects, used in the linetype legend. |
species |
The species to be selected. Optional. By default all target species are selected. A vector of species names, or a numeric vector with the species indices, or a logical vector indicating for each species whether it is to be selected (TRUE) or not. |
wlim |
A numeric vector of length two providing lower and upper limits
for the w axis. Use NA for the default: the lower default is
|
llim |
A numeric vector of length two providing lower and upper limits
for the length axis when |
ylim |
A numeric vector of length two providing lower and upper limits
for the y axis. Use NA to auto-scale to the data range. Values below 1e-20
are always filtered out from the data regardless of |
power |
The abundance is plotted as the number density times the weight
raised to |
biomass |
|
total |
A boolean value that determines whether the total over all species in the system is plotted as well. Note that even if the plot only shows a selection of species, the total is including all species. Default is FALSE. |
resource |
A boolean value that determines whether resource is included. Default is FALSE. |
background |
A boolean value that determines whether background species are included. Ignored if the model does not contain background species. Default is TRUE. |
highlight |
Name or vector of names of the species to be highlighted by being plotted with thicker lines. |
normalise |
If |
log_x |
If |
log_y |
If |
log |
Character string specifying which axes should use a log10 scale,
in the same form as the base |
size_axis |
Whether to plot size as weight ( |
... |
Additional arguments passed to |
Details
plotlyCDF2() is the interactive plotly version.
Value
A ggplot2 object. plotlyCDF2() returns a plotly object.
See Also
Other plotting functions:
addPlot(),
animate(),
plot,
plot2(),
plotBiomass(),
plotCDF(),
plotDiet(),
plotFMort(),
plotFeedingLevel(),
plotGrowthCurves(),
plotMizerParams,
plotMizerSim,
plotPredMort(),
plotRelative(),
plotSpectra(),
plotSpectra2(),
plotSpectraRelative(),
plotYield(),
plotYieldGear(),
plotting_functions
Examples
sim1 <- project(NS_params, t_max = 10, progress_bar = FALSE)
sim2 <- project(NS_params, effort = 0.5, t_max = 10, progress_bar = FALSE)
plotCDF2(sim1, sim2, "Original", "Effort = 0.5")
Make a plot comparing two data frames
Description
Used internally by the comparison plotting functions such as plotSpectra2()
and plotCDF2(). The two data frames are combined and drawn with colour
identifying the species or group and linetype identifying the object.
Usage
plotComparisonDataFrame(
frame1,
frame2,
params,
name1 = "First",
name2 = "Second",
xlab = waiver(),
ylab = waiver(),
xtrans = "identity",
ytrans = "identity",
xlim = c(NA, NA),
ylim = c(NA, NA),
y_ticks = 6,
highlight = NULL,
legend_var = "Legend",
size_axis = NULL
)
Arguments
frame1, frame2 |
Data frames sharing the same first three variables (x, y
and grouping variable). The names of |
params |
A MizerParams object, used for the line colours. |
name1, name2 |
Labels for the two data frames, used in the linetype legend. |
xlab, ylab |
Labels for the x and y axes. |
xtrans, ytrans |
Transformations for the x and y axes, e.g. |
xlim, ylim |
Numeric vectors of length two giving the axis limits. Use
|
y_ticks |
The approximate number of ticks desired on the y axis. |
highlight |
Name or vector of names of the species to be highlighted. |
legend_var |
Name of the variable used in the legend and to determine the line colour. |
size_axis |
Optional. If non-NULL, the x-axis is converted to weight
( |
Value
A mizer_plot (ggplot2) object.
Make a plot from a data frame
Description
This is used internally by most plotting functions.
Usage
plotDataFrame(
frame,
params,
style = "line",
xlab = waiver(),
ylab = waiver(),
xtrans = "identity",
ytrans = "identity",
xlim = c(NA, NA),
ylim = c(NA, NA),
y_ticks = 6,
highlight = NULL,
legend_var = NULL,
wrap_var = NULL,
wrap_scale = NULL
)
Arguments
frame |
A data frame with at least three variables. The first three variables are used, in that order, as:
|
params |
A MizerParams object, which is used for the line colours and line types. |
style |
The style of the plot. Available options are |
xlab |
Label for the x-axis |
ylab |
Label for the y-axis |
xtrans |
Transformation for the x-axis. Often "log10" may be useful instead of the default of "identity". |
ytrans |
Transformation for the y-axis. |
xlim |
A numeric vector of length two providing lower and upper limits for the x axis. Use NA to refer to the existing minimum or maximum. |
ylim |
A numeric vector of length two providing lower and upper limits for the y axis. Use NA to refer to the existing minimum or maximum. |
y_ticks |
The approximate number of ticks desired on the y axis |
highlight |
Name or vector of names of the species to be highlighted. |
legend_var |
The name of the variable that should be used in the legend and to determine the line style. If NULL then the grouping variable is used for this purpose. |
wrap_var |
Optional. The name of the variable that should be used for creating wrapped facets. |
wrap_scale |
Optional. Used to pass the scales argument to facet_wrap(). |
Value
A ggplot2 object
Plot diet, resolved by prey species, as function of predator at size.
Description
Plots the proportions with which each
prey species contributes to the total biomass consumed by the specified
predator species, as a function of the predator's size. These proportions are
obtained with getDiet().
Usage
plotDiet(
object,
species = NULL,
wlim = c(NA, NA),
llim = c(NA, NA),
size_axis = c("w", "l"),
return_data = FALSE,
log_x = TRUE,
log_y = FALSE,
log = NULL,
...
)
Arguments
object |
An object of class MizerSim or MizerParams. |
species |
The species to be selected. Optional. By default all target species are selected. A vector of species names, or a numeric vector with the species indices, or a logical vector indicating for each species whether it is to be selected (TRUE) or not. |
wlim |
A numeric vector of length two providing lower and upper limits
for the weight (x) axis. Use |
llim |
A numeric vector of length two providing lower and upper limits
for the length (x) axis when |
size_axis |
Whether to plot size as weight ( |
return_data |
A boolean value that determines whether the formatted data used for the plot is returned instead of the plot itself. Default is FALSE. |
log_x |
If |
log_y |
If |
log |
Character string specifying which axes should use log10 scales,
in the same form as the base |
... |
Further arguments used by only some of the methods: For
|
Details
Prey species that contribute less than 1 permille to the diet are suppressed in the plot. The plot only extends to predator sizes where the predator has a meaningful abundance (defined as having a biomass density greater than 0.1% of its maximum biomass density).
If more than one predator species is selected, then the plot contains one facet for each species.
Value
A ggplot2 object, unless return_data = TRUE, in which case a data
frame with the four variables 'Predator', 'w' (or 'l' if
size_axis = "l"), 'Proportion', 'Prey' is returned.
plotlyDiet() returns a plotly object.
See Also
Other plotting functions:
addPlot(),
animate(),
plot,
plot2(),
plotBiomass(),
plotCDF(),
plotCDF2(),
plotFMort(),
plotFeedingLevel(),
plotGrowthCurves(),
plotMizerParams,
plotMizerSim,
plotPredMort(),
plotRelative(),
plotSpectra(),
plotSpectra2(),
plotSpectraRelative(),
plotYield(),
plotYieldGear(),
plotting_functions
Examples
plotDiet(NS_params, species = "Cod")
plotDiet(NS_params, species = 5:9)
# Returning the data frame
fr <- plotDiet(NS_params, species = "Cod", return_data = TRUE)
str(fr)
Plot total fishing mortality of each species by size
Description
After running a projection, plot the total fishing mortality of each species by size. The total fishing mortality is averaged over the specified time range (a single value for the time range can be used to plot a single time step).
Usage
plotFMort(
object,
species = NULL,
all.sizes = FALSE,
highlight = NULL,
wlim = c(NA, NA),
llim = c(NA, NA),
size_axis = c("w", "l"),
return_data = FALSE,
log_x = TRUE,
log_y = FALSE,
log = NULL,
...
)
Arguments
object |
An object of class MizerSim or MizerParams. |
species |
The species to be selected. Optional. By default all target species are selected. A vector of species names, or a numeric vector with the species indices, or a logical vector indicating for each species whether it is to be selected (TRUE) or not. |
all.sizes |
If TRUE, then fishing mortality is plotted also for sizes outside a species' size range. Default FALSE. |
highlight |
Name or vector of names of the species to be highlighted. |
wlim |
A numeric vector of length two providing lower and upper limits
for the weight (x) axis. Use |
llim |
A numeric vector of length two providing lower and upper limits
for the length (x) axis when |
size_axis |
Whether to plot size as weight ( |
return_data |
A boolean value that determines whether the formatted data used for the plot is returned instead of the plot itself. Default is FALSE. |
log_x |
If |
log_y |
If |
log |
Character string specifying which axes should use log10 scales,
in the same form as the base |
... |
Further arguments used by only some of the methods: For
|
Value
A ggplot2 object, unless return_data = TRUE, in which case a data
frame with the three variables 'w' (or 'l' if size_axis = "l"), 'value',
'Species' is returned.
See Also
plotting_functions, getFMort()
Other plotting functions:
addPlot(),
animate(),
plot,
plot2(),
plotBiomass(),
plotCDF(),
plotCDF2(),
plotDiet(),
plotFeedingLevel(),
plotGrowthCurves(),
plotMizerParams,
plotMizerSim,
plotPredMort(),
plotRelative(),
plotSpectra(),
plotSpectra2(),
plotSpectraRelative(),
plotYield(),
plotYieldGear(),
plotting_functions
Examples
params <- NS_params
sim <- project(params, effort=1, t_max=20, t_save = 2, progress_bar = FALSE)
plotFMort(sim)
plotFMort(sim, highlight = c("Cod", "Haddock"))
# Returning the data frame
fr <- plotFMort(sim, return_data = TRUE)
str(fr)
Plot the feeding level of species by size
Description
After running a projection, plot the feeding level of each species by size. The feeding level is averaged over the specified time range (a single value for the time range can be used).
Usage
plotFeedingLevel(
object,
species = NULL,
all.sizes = FALSE,
highlight = NULL,
include_critical = FALSE,
wlim = c(NA, NA),
llim = c(NA, NA),
size_axis = c("w", "l"),
return_data = FALSE,
log_x = TRUE,
log_y = FALSE,
log = NULL,
...
)
Arguments
object |
An object of class MizerSim or MizerParams. |
species |
The species to be selected. Optional. By default all target species are selected. A vector of species names, or a numeric vector with the species indices, or a logical vector indicating for each species whether it is to be selected (TRUE) or not. |
all.sizes |
If TRUE, then feeding level is plotted also for sizes outside a species' size range. Default FALSE. |
highlight |
Name or vector of names of the species to be highlighted. |
include_critical |
If TRUE, then the critical feeding level is also plotted. Default FALSE. |
wlim |
A numeric vector of length two providing lower and upper limits
for the weight (x) axis. Use |
llim |
A numeric vector of length two providing lower and upper limits
for the length (x) axis when |
size_axis |
Whether to plot size as weight ( |
return_data |
A boolean value that determines whether the formatted data used for the plot is returned instead of the plot itself. Default is FALSE. |
log_x |
If |
log_y |
If |
log |
Character string specifying which axes should use log10 scales,
in the same form as the base |
... |
Further arguments used by only some of the methods: For
|
Details
When called with a MizerSim object, the feeding level is averaged over the specified time range (a single value for the time range can be used to plot a single time step). When called with a MizerParams object the initial feeding level is plotted.
If include_critical = TRUE then the critical feeding level (the feeding
level at which the intake just covers the metabolic cost) is also plotted,
with a thinner line. This line should always stay below the line of the
actual feeding level, because the species would stop growing at any point
where the feeding level drops to the critical feeding level.
Value
A ggplot2 object, unless return_data = TRUE, in which case a data
frame with the variables 'w' (or 'l' if size_axis = "l"), 'value' and
'Species' is returned. If also include_critical = TRUE then the data
frame contains a fourth variable 'Type' that distinguishes between
'actual' and 'critical' feeding level.
See Also
plotting_functions, getFeedingLevel()
Other plotting functions:
addPlot(),
animate(),
plot,
plot2(),
plotBiomass(),
plotCDF(),
plotCDF2(),
plotDiet(),
plotFMort(),
plotGrowthCurves(),
plotMizerParams,
plotMizerSim,
plotPredMort(),
plotRelative(),
plotSpectra(),
plotSpectra2(),
plotSpectraRelative(),
plotYield(),
plotYieldGear(),
plotting_functions
Examples
params <- NS_params
sim <- project(params, effort=1, t_max=20, t_save = 2, progress_bar = FALSE)
plotFeedingLevel(sim)
plotFeedingLevel(sim, time_range = 10:20, species = c("Cod", "Herring"),
include_critical = TRUE)
# Returning the data frame
fr <- plotFeedingLevel(sim, return_data = TRUE)
str(fr)
Plot growth curves
Description
The growth curves represent the average age of all the living fish of a
species as a function of their size. So it would be natural to plot size
on the x-axis. But to follow the usual convention from age-based models, we
plot size on the y-axis and age on the x-axis.
Usage
plotGrowthCurves(
object,
species = NULL,
max_age = 20,
percentage = FALSE,
species_panel = FALSE,
highlight = NULL,
size_at_age = NULL,
return_data = FALSE,
log_x = FALSE,
log_y = FALSE,
log = NULL,
...
)
Arguments
object |
An object of class MizerSim or MizerParams. |
species |
The species to be selected. Optional. By default all target species are selected. A vector of species names, or a numeric vector with the species indices, or a logical vector indicating for each species whether it is to be selected (TRUE) or not. |
max_age |
The age up to which to run the growth curve. Default is 20. |
percentage |
Boolean value. If TRUE, the size is given as a percentage of the maximal size. |
species_panel |
If TRUE (default), and |
highlight |
Name or vector of names of the species to be highlighted. |
size_at_age |
A data frame with observed size at age data to be plotted
on top of growth curve graphs. Should contain columns |
return_data |
A boolean value that determines whether the formatted data used for the plot is returned instead of the plot itself. Default is FALSE. |
log_x |
If |
log_y |
If |
log |
Character string specifying which axes should use log10 scales,
in the same form as the base |
... |
Unused. |
Details
In each panel for a single species, a horizontal line is included that indicate the maturity size of the species and a vertical line indicating its maturity age.
If size at age data is passed via the size_at_age argument, this is plotted
on top of the growth curve. When comparing this to the growth curves, you
need to remember that the growth curves should only represent the average
age at each size. So a scatter in the x-direction around the curve is to be
expected.
If the species parameters contain the variables a and b for length to
weight conversion and the von Bertalanffy parameter k_vb, w_inf (and
optionally t0), then the von Bertalanffy growth curve is superimposed in
black. Note that the von Bertalanffy curve (which approximates the average
length at each age) should not be compared directly to the mizer growth
curves (which approximate the average age at each length).
Value
A ggplot2 object
See Also
Other plotting functions:
addPlot(),
animate(),
plot,
plot2(),
plotBiomass(),
plotCDF(),
plotCDF2(),
plotDiet(),
plotFMort(),
plotFeedingLevel(),
plotMizerParams,
plotMizerSim,
plotPredMort(),
plotRelative(),
plotSpectra(),
plotSpectra2(),
plotSpectraRelative(),
plotYield(),
plotYieldGear(),
plotting_functions
Examples
params <- NS_params
sim <- project(params, effort=1, t_max=20, t_save = 2, progress_bar = FALSE)
plotGrowthCurves(sim, percentage = TRUE)
plotGrowthCurves(sim, species = "Cod", max_age = 24)
plotGrowthCurves(sim, species_panel = TRUE)
# Returning the data frame
fr <- plotGrowthCurves(sim, return_data = TRUE)
str(fr)
Create a hover-enabled plotly plot from a mizer object
Description
Creates an interactive plotly version of a mizer plot. Can be called on any
mizer array object (such as those returned by getEncounter(),
getBiomass(), etc.) or on any mizer_plot object returned by the named
plot functions such as plotBiomass(), plotSpectra(), etc.
Usage
## S3 method for class 'ArrayTimeBySpecies'
plotHover(x, ...)
plotHover(x, ...)
Arguments
x |
A |
... |
Arguments passed to the corresponding |
Value
A plotly object.
See Also
plot(), plotBiomass(), plotSpectra(), plotting_functions
Examples
plotHover(getEncounter(NS_params))
plotHover(getBiomass(NS_sim))
plotHover(getFMort(NS_sim))
plotHover(getResourceMort(NS_params))
plotHover(NResource(NS_sim))
Alias for plotPredMort()
Description
An alias provided for backward compatibility with mizer version <= 1.0
Usage
plotM2(
object,
species = NULL,
all.sizes = FALSE,
highlight = NULL,
wlim = c(NA, NA),
llim = c(NA, NA),
size_axis = c("w", "l"),
return_data = FALSE,
log_x = TRUE,
log_y = FALSE,
log = NULL,
...
)
Arguments
object |
An object of class MizerSim or MizerParams. |
species |
The species to be selected. Optional. By default all target species are selected. A vector of species names, or a numeric vector with the species indices, or a logical vector indicating for each species whether it is to be selected (TRUE) or not. |
all.sizes |
If TRUE, then predation mortality is plotted also for sizes outside a species' size range. Default FALSE. |
highlight |
Name or vector of names of the species to be highlighted. |
wlim |
A numeric vector of length two providing lower and upper limits
for the weight (x) axis. Use |
llim |
A numeric vector of length two providing lower and upper limits
for the length (x) axis when |
size_axis |
Whether to plot size as weight ( |
return_data |
A boolean value that determines whether the formatted data used for the plot is returned instead of the plot itself. Default is FALSE. |
log_x |
If |
log_y |
If |
log |
Character string specifying which axes should use log10 scales,
in the same form as the base |
... |
Further arguments used by only some of the methods: For
|
Value
A ggplot2 object, unless return_data = TRUE, in which case a data
frame with the three variables 'w' (or 'l' if size_axis = "l"), 'value',
'Species' is returned.
See Also
plotting_functions, getPredMort()
Other plotting functions:
addPlot(),
animate(),
plot,
plot2(),
plotBiomass(),
plotCDF(),
plotCDF2(),
plotDiet(),
plotFMort(),
plotFeedingLevel(),
plotGrowthCurves(),
plotMizerParams,
plotMizerSim,
plotRelative(),
plotSpectra(),
plotSpectra2(),
plotSpectraRelative(),
plotYield(),
plotYieldGear(),
plotting_functions
Examples
params <- NS_params
sim <- project(params, effort=1, t_max=20, t_save = 2, progress_bar = FALSE)
plotPredMort(sim)
plotPredMort(sim, time_range = 10:20)
# Returning the data frame
fr <- plotPredMort(sim, return_data = TRUE)
str(fr)
Summary plot for MizerParams objects
Description
Produces 3 plots in the same window: abundance spectra, feeding
level and predation mortality of each species against size. This method just
puts the plots generated by plotFeedingLevel(), plotPredMort() and
plotSpectra() all in one window.
Usage
## S3 method for class 'MizerParams'
plot(x, ...)
Arguments
x |
An object of class MizerParams |
... |
Arguments passed on to the individual plotting functions
|
Value
A viewport object
See Also
Other plotting functions:
addPlot(),
animate(),
plot,
plot2(),
plotBiomass(),
plotCDF(),
plotCDF2(),
plotDiet(),
plotFMort(),
plotFeedingLevel(),
plotGrowthCurves(),
plotMizerSim,
plotPredMort(),
plotRelative(),
plotSpectra(),
plotSpectra2(),
plotSpectraRelative(),
plotYield(),
plotYieldGear(),
plotting_functions
Examples
params <- NS_params
plot(params)
Summary plot for MizerSim objects
Description
After running a projection, produces 5 plots in the same window: feeding
level, abundance spectra, predation mortality and fishing mortality of each
species by size; and biomass of each species through time. This method just
puts the plots generated by plotBiomass(), plotFeedingLevel(),
plotSpectra(), plotPredMort() and plotFMort() all in one window.
Usage
## S3 method for class 'MizerSim'
plot(x, ...)
Arguments
x |
An object of class MizerSim |
... |
Arguments passed on to the individual plotting functions
|
Value
A viewport object
See Also
Other plotting functions:
addPlot(),
animate(),
plot,
plot2(),
plotBiomass(),
plotCDF(),
plotCDF2(),
plotDiet(),
plotFMort(),
plotFeedingLevel(),
plotGrowthCurves(),
plotMizerParams,
plotPredMort(),
plotRelative(),
plotSpectra(),
plotSpectra2(),
plotSpectraRelative(),
plotYield(),
plotYieldGear(),
plotting_functions
Examples
params <- NS_params
sim <- project(params, effort=1, t_max=20, t_save = 2, progress_bar = FALSE)
plot(sim)
Plot predation mortality rate of each species against size
Description
After running a projection, plot the predation mortality rate of each species by size. The mortality rate is averaged over the specified time range (a single value for the time range can be used to plot a single time step).
Usage
plotPredMort(
object,
species = NULL,
all.sizes = FALSE,
highlight = NULL,
wlim = c(NA, NA),
llim = c(NA, NA),
size_axis = c("w", "l"),
return_data = FALSE,
log_x = TRUE,
log_y = FALSE,
log = NULL,
...
)
Arguments
object |
An object of class MizerSim or MizerParams. |
species |
The species to be selected. Optional. By default all target species are selected. A vector of species names, or a numeric vector with the species indices, or a logical vector indicating for each species whether it is to be selected (TRUE) or not. |
all.sizes |
If TRUE, then predation mortality is plotted also for sizes outside a species' size range. Default FALSE. |
highlight |
Name or vector of names of the species to be highlighted. |
wlim |
A numeric vector of length two providing lower and upper limits
for the weight (x) axis. Use |
llim |
A numeric vector of length two providing lower and upper limits
for the length (x) axis when |
size_axis |
Whether to plot size as weight ( |
return_data |
A boolean value that determines whether the formatted data used for the plot is returned instead of the plot itself. Default is FALSE. |
log_x |
If |
log_y |
If |
log |
Character string specifying which axes should use log10 scales,
in the same form as the base |
... |
Further arguments used by only some of the methods: For
|
Value
A ggplot2 object, unless return_data = TRUE, in which case a data
frame with the three variables 'w' (or 'l' if size_axis = "l"), 'value',
'Species' is returned.
See Also
plotting_functions, getPredMort()
Other plotting functions:
addPlot(),
animate(),
plot,
plot2(),
plotBiomass(),
plotCDF(),
plotCDF2(),
plotDiet(),
plotFMort(),
plotFeedingLevel(),
plotGrowthCurves(),
plotMizerParams,
plotMizerSim,
plotRelative(),
plotSpectra(),
plotSpectra2(),
plotSpectraRelative(),
plotYield(),
plotYieldGear(),
plotting_functions
Examples
params <- NS_params
sim <- project(params, effort=1, t_max=20, t_save = 2, progress_bar = FALSE)
plotPredMort(sim)
plotPredMort(sim, time_range = 10:20)
# Returning the data frame
fr <- plotPredMort(sim, return_data = TRUE)
str(fr)
Plot relative difference between two mizer arrays
Description
plotRelative() plots the difference between two compatible mizer array
objects relative to their average. If the values in the first object are
N_1 and the values in the second are N_2, it plots
2 (N_2 - N_1) / (N_1 + N_2).
Usage
plotRelative(
x,
y,
species = NULL,
log_x,
ylim = c(NA, NA),
total = FALSE,
background = TRUE,
...
)
Arguments
x |
The first of two compatible mizer array objects to compare.
Can be an |
y |
The second mizer array object, compatible with |
species |
Character vector of species to include. |
log_x |
If |
ylim |
A numeric vector of length two providing lower and upper limits for the value (y) axis. |
total |
A boolean value that determines whether the total over all
selected species is plotted as well. Default is |
background |
A boolean value that determines whether background species
are included. Ignored if the model does not contain background species.
Default is |
... |
Further arguments used by only some of the methods: For
For
For
|
Value
A ggplot2 object.
See Also
Other plotting functions:
addPlot(),
animate(),
plot,
plot2(),
plotBiomass(),
plotCDF(),
plotCDF2(),
plotDiet(),
plotFMort(),
plotFeedingLevel(),
plotGrowthCurves(),
plotMizerParams,
plotMizerSim,
plotPredMort(),
plotSpectra(),
plotSpectra2(),
plotSpectraRelative(),
plotYield(),
plotYieldGear(),
plotting_functions
Examples
params <- NS_params
given_species_params(params)["Cod", "w_mat"] <- 1200
plotRelative(getEGrowth(NS_params), getEGrowth(params),
wlim = c(500, 2000), log_x = FALSE, species = "Cod")
Make a plot of the relative difference between two data frames
Description
Used internally by plotSpectraRelative() and similar functions. The two
data frames are joined on their shared variables and the relative difference
of their y-values is plotted against the x-variable.
Usage
plotRelativeDataFrame(
frame1,
frame2,
params,
xlab = waiver(),
xtrans = "identity",
xlim = c(NA, NA),
ylim = c(NA, NA),
highlight = NULL,
legend_var = "Legend",
size_axis = NULL
)
Arguments
frame1, frame2 |
Data frames sharing the same first three variables (x, y
and grouping variable). The names of |
params |
A MizerParams object, used for the line colours. |
xlab |
Label for the x-axis. |
xtrans |
Transformation for the x-axis, e.g. |
xlim, ylim |
Numeric vectors of length two giving the axis limits. Use
|
highlight |
Name or vector of names of the species to be highlighted. |
legend_var |
Name of the variable used in the legend and to determine the line colour. |
size_axis |
Optional. If non-NULL, the x-axis is converted to weight
( |
Value
A mizer_plot (ggplot2) object showing the relative difference.
Plot abundance and biomass spectra
Description
plotSpectra() plots the number density multiplied by a power of the
weight, with the power specified by the power argument. When called with a
MizerSim object, the abundance is averaged over the specified time range
(a single value for the time range can be used to plot a single time step).
When called with a MizerParams object the initial abundance is plotted.
Usage
plotSpectra(
object,
species = NULL,
wlim = c(NA, NA),
llim = c(NA, NA),
ylim = c(NA, NA),
power = 1,
biomass = TRUE,
total = FALSE,
resource = TRUE,
background = TRUE,
highlight = NULL,
log_x = TRUE,
log_y = TRUE,
log = NULL,
size_axis = c("w", "l"),
return_data = FALSE,
...
)
Arguments
object |
An object of class MizerSim or MizerParams. |
species |
The species to be selected. Optional. By default all target species are selected. A vector of species names, or a numeric vector with the species indices, or a logical vector indicating for each species whether it is to be selected (TRUE) or not. |
wlim |
A numeric vector of length two providing lower and upper limits
for the w axis. Use NA for the default: the lower default is
|
llim |
A numeric vector of length two providing lower and upper limits
for the length axis when |
ylim |
A numeric vector of length two providing lower and upper limits
for the y axis. Use NA to auto-scale to the data range. Values below 1e-20
are always filtered out from the data regardless of |
power |
The abundance is plotted as the number density times the weight
raised to |
biomass |
|
total |
A boolean value that determines whether the total over all species in the system is plotted as well. Note that even if the plot only shows a selection of species, the total is including all species. Default is FALSE. |
resource |
A boolean value that determines whether resource is included. Default is TRUE. |
background |
A boolean value that determines whether background species are included. Ignored if the model does not contain background species. Default is TRUE. |
highlight |
Name or vector of names of the species to be highlighted by being plotted with thicker lines. |
log_x |
If |
log_y |
If |
log |
Character string specifying which axes should use log10 scales,
in the same form as the base |
size_axis |
Whether to plot size as weight ( |
return_data |
A boolean value that determines whether the formatted data used for the plot is returned instead of the plot itself. Default value is FALSE |
... |
Further arguments used by only some of the methods: For
|
Details
plotlySpectra() is the interactive plotly version. To compare spectra from
two objects use plotSpectra2(). To show relative differences use
plotSpectraRelative().
Value
A ggplot2 object, unless return_data = TRUE, in which case a data
frame with the four variables 'w' (or 'l' if size_axis = "l"), 'value',
'Species', 'Legend' is returned. plotlySpectra() returns a plotly object.
See Also
Other plotting functions:
addPlot(),
animate(),
plot,
plot2(),
plotBiomass(),
plotCDF(),
plotCDF2(),
plotDiet(),
plotFMort(),
plotFeedingLevel(),
plotGrowthCurves(),
plotMizerParams,
plotMizerSim,
plotPredMort(),
plotRelative(),
plotSpectra2(),
plotSpectraRelative(),
plotYield(),
plotYieldGear(),
plotting_functions
Examples
params <- NS_params
sim <- project(params, effort=1, t_max=20, t_save = 2, progress_bar = FALSE)
plotSpectra(sim)
plotSpectra(sim, wlim = c(1e-6, NA))
plotSpectra(sim, time_range = 10:20)
plotSpectra(sim, time_range = 10:20, power = 0)
plotSpectra(sim, species = c("Cod", "Herring"), power = 1)
plotSpectra(sim, species = c("Cod", "Herring"), size_axis = "l")
# Returning the data frame
fr <- plotSpectra(sim, return_data = TRUE)
str(fr)
Compare abundance and biomass spectra from two objects
Description
plotSpectra2() compares the abundance spectra from two MizerParams or
MizerSim objects in a single plot. Colours identify species or groups and
linetype identifies the object.
Usage
plotSpectra2(
object1,
object2,
name1 = "First",
name2 = "Second",
species = NULL,
wlim = c(NA, NA),
llim = c(NA, NA),
ylim = c(NA, NA),
power = 1,
biomass = TRUE,
total = FALSE,
resource = TRUE,
background = TRUE,
highlight = NULL,
log_x = TRUE,
log_y = TRUE,
log = NULL,
size_axis = c("w", "l"),
...
)
Arguments
object1 |
First |
object2 |
Second |
name1, name2 |
Labels for the two objects, used in the linetype legend. |
species |
The species to be selected. Optional. By default all target species are selected. A vector of species names, or a numeric vector with the species indices, or a logical vector indicating for each species whether it is to be selected (TRUE) or not. |
wlim |
A numeric vector of length two providing lower and upper limits
for the w axis. Use NA for the default: the lower default is
|
llim |
A numeric vector of length two providing lower and upper limits
for the length axis when |
ylim |
A numeric vector of length two providing lower and upper limits
for the y axis. Use NA to auto-scale to the data range. Values below 1e-20
are always filtered out from the data regardless of |
power |
The abundance is plotted as the number density times the weight
raised to |
biomass |
|
total |
A boolean value that determines whether the total over all species in the system is plotted as well. Note that even if the plot only shows a selection of species, the total is including all species. Default is FALSE. |
resource |
A boolean value that determines whether resource is included. Default is TRUE. |
background |
A boolean value that determines whether background species are included. Ignored if the model does not contain background species. Default is TRUE. |
highlight |
Name or vector of names of the species to be highlighted by being plotted with thicker lines. |
log_x |
If |
log_y |
If |
log |
Character string specifying which axes should use log10 scales,
in the same form as the base |
size_axis |
Whether to plot size as weight ( |
... |
Additional arguments passed to |
Details
plotlySpectra2() is the interactive plotly version.
Value
A ggplot2 object. plotlySpectra2() returns a plotly object.
See Also
plotting_functions, plotSpectra(), plotSpectraRelative()
Other plotting functions:
addPlot(),
animate(),
plot,
plot2(),
plotBiomass(),
plotCDF(),
plotCDF2(),
plotDiet(),
plotFMort(),
plotFeedingLevel(),
plotGrowthCurves(),
plotMizerParams,
plotMizerSim,
plotPredMort(),
plotRelative(),
plotSpectra(),
plotSpectraRelative(),
plotYield(),
plotYieldGear(),
plotting_functions
Examples
sim1 <- project(NS_params, t_max = 10, progress_bar = FALSE)
sim2 <- project(NS_params, effort = 0.5, t_max = 10, progress_bar = FALSE)
plotSpectra2(sim1, sim2, "Original", "Effort = 0.5")
Plot relative difference between abundance spectra
Description
plotSpectraRelative() plots the difference between the spectra relative to
their average. If we denote the number density from the first object as
N_1(w) and that from the second object as N_2(w), then this
plot shows
2 (N_2(w) - N_1(w)) / (N_2(w) + N_1(w)).
Note that it does not matter whether the relative difference is calculated
for number density, biomass density, or biomass density in log weight,
because the factors of w by which the densities differ cancel out in
the relative difference.
Usage
plotSpectraRelative(
object1,
object2,
species = NULL,
wlim = c(NA, NA),
llim = c(NA, NA),
ylim = c(NA, NA),
total = FALSE,
resource = TRUE,
background = TRUE,
highlight = NULL,
log_x = TRUE,
size_axis = c("w", "l"),
...
)
Arguments
object1 |
First |
object2 |
Second |
species |
The species to be selected. Optional. By default all target species are selected. A vector of species names, or a numeric vector with the species indices, or a logical vector indicating for each species whether it is to be selected (TRUE) or not. |
wlim |
A numeric vector of length two providing lower and upper limits
for the w axis. Use NA for the default: the lower default is
|
llim |
A numeric vector of length two providing lower and upper limits
for the length axis when |
ylim |
A numeric vector of length two providing lower and upper limits
for the y axis (the relative difference). Use |
total |
A boolean value that determines whether the total over all species in the system is plotted as well. Note that even if the plot only shows a selection of species, the total is including all species. Default is FALSE. |
resource |
A boolean value that determines whether resource is included. Default is TRUE. |
background |
A boolean value that determines whether background species are included. Ignored if the model does not contain background species. Default is TRUE. |
highlight |
Name or vector of names of the species to be highlighted by being plotted with thicker lines. |
log_x |
If |
size_axis |
Whether to plot size as weight ( |
... |
Additional arguments passed to |
Details
plotlySpectraRelative() is the interactive plotly version.
Value
A ggplot2 object. plotlySpectraRelative() returns a plotly object.
See Also
plotting_functions, plotSpectra(), plotSpectra2()
Other plotting functions:
addPlot(),
animate(),
plot,
plot2(),
plotBiomass(),
plotCDF(),
plotCDF2(),
plotDiet(),
plotFMort(),
plotFeedingLevel(),
plotGrowthCurves(),
plotMizerParams,
plotMizerSim,
plotPredMort(),
plotRelative(),
plotSpectra(),
plotSpectra2(),
plotYield(),
plotYieldGear(),
plotting_functions
Examples
sim1 <- project(NS_params, t_max = 10, progress_bar = FALSE)
sim2 <- project(NS_params, effort = 0.5, t_max = 10, progress_bar = FALSE)
plotSpectraRelative(sim1, sim2)
Plot the total yield of species through time
Description
After running a projection, the total yield of each species across all
fishing gears can be plotted against time. The yield is obtained with
getYield().
Usage
plotYield(
object,
sim2 = NULL,
species = NULL,
total = FALSE,
log_x = FALSE,
log_y = TRUE,
log = NULL,
ylim = c(NA, NA),
tlim = c(NA, NA),
highlight = NULL,
return_data = FALSE,
...
)
Arguments
object |
An object of class MizerSim |
sim2 |
An optional second object of class MizerSim. If this is provided its yields will be shown on the same plot in bolder lines. |
species |
The species to be selected. Optional. By default all target species are selected. A vector of species names, or a numeric vector with the species indices, or a logical vector indicating for each species whether it is to be selected (TRUE) or not. |
total |
A boolean value that determines whether the total yield from all species is plotted as well. Default is FALSE. |
log_x |
If |
log_y |
If |
log |
Character string specifying which axes should use log10 scales,
in the same form as the base |
ylim |
A numeric vector of length two providing lower and upper limits
for the y axis. Use |
tlim |
A numeric vector of length two providing lower and upper limits
for the time axis, e.g. |
highlight |
Name or vector of names of the species to be highlighted. |
return_data |
A boolean value that determines whether the formatted data used for the plot is returned instead of the plot itself. Default is FALSE. |
... |
Arguments passed to |
Value
A ggplot2 object, unless return_data = TRUE, in which case a data
frame with the three variables 'Year', 'Yield', 'Species' is returned.
See Also
plotting_functions, getYield()
Other plotting functions:
addPlot(),
animate(),
plot,
plot2(),
plotBiomass(),
plotCDF(),
plotCDF2(),
plotDiet(),
plotFMort(),
plotFeedingLevel(),
plotGrowthCurves(),
plotMizerParams,
plotMizerSim,
plotPredMort(),
plotRelative(),
plotSpectra(),
plotSpectra2(),
plotSpectraRelative(),
plotYieldGear(),
plotting_functions
Examples
params <- NS_params
sim <- project(params, effort = 1, t_max = 20, t_save = 0.2, progress_bar = FALSE)
plotYield(sim)
plotYield(sim, species = c("Cod", "Herring"), total = TRUE)
# Comparing with yield from twice the effort
sim2 <- project(params, effort=2, t_max=20, t_save = 0.2, progress_bar = FALSE)
plotYield(sim, sim2, species = c("Cod", "Herring"), log = FALSE)
# Returning the data frame
fr <- plotYield(sim, return_data = TRUE)
str(fr)
Plot the total yield of each species by gear through time
Description
After running a projection, the total yield of each species by fishing gear can be plotted against time.
Usage
plotYieldGear(
object,
species = NULL,
gears = NULL,
total = FALSE,
log_x = FALSE,
log_y = TRUE,
log = NULL,
ylim = c(NA, NA),
tlim = c(NA, NA),
highlight = NULL,
return_data = FALSE,
...
)
Arguments
object |
An object of class MizerSim |
species |
The species to be selected. Optional. By default all target species are selected. A vector of species names, or a numeric vector with the species indices, or a logical vector indicating for each species whether it is to be selected (TRUE) or not. |
gears |
A vector of gear names to be included in the plot. Default is all gears. |
total |
A boolean value that determines whether the total yield from all species is plotted as well. Default is FALSE. |
log_x |
If |
log_y |
If |
log |
Character string specifying which axes should use log10 scales,
in the same form as the base |
ylim |
A numeric vector of length two providing lower and upper limits
for the y axis. Use |
tlim |
A numeric vector of length two providing lower and upper limits
for the time axis, e.g. |
highlight |
Name or vector of names of the species to be highlighted. |
return_data |
A boolean value that determines whether the formatted data used for the plot is returned instead of the plot itself. Default is FALSE. |
... |
Arguments passed to |
Details
This plot is pretty easy to do by hand. It just
gets the biomass using the getYieldGear() method and plots using
the ggplot2 package. You can then fiddle about with colours and linetypes
etc. Just look at the source code for details.
Value
A ggplot2 object, unless return_data = TRUE, in which case a data
frame with the four variables 'Year', 'Yield', 'Species' and 'Gear' is
returned.
See Also
plotting_functions, getYieldGear()
Other plotting functions:
addPlot(),
animate(),
plot,
plot2(),
plotBiomass(),
plotCDF(),
plotCDF2(),
plotDiet(),
plotFMort(),
plotFeedingLevel(),
plotGrowthCurves(),
plotMizerParams,
plotMizerSim,
plotPredMort(),
plotRelative(),
plotSpectra(),
plotSpectra2(),
plotSpectraRelative(),
plotYield(),
plotting_functions
Examples
params <- NS_params
sim <- project(params, effort=1, t_max=20, t_save = 0.2, progress_bar = FALSE)
plotYieldGear(sim)
plotYieldGear(sim, species = c("Cod", "Herring"), total = TRUE)
# Returning the data frame
fr <- plotYieldGear(sim, return_data = TRUE)
str(fr)
Plotting observed vs. model yields
Description
If yield observations are available for at least some species via the
yield_observed column in the species parameter data frame, this function
plots the yield of each species in the model against the observed
yields. When called with a MizerSim object, the plot will use the model
yields predicted for the final time step in the simulation.
Usage
plotYieldObservedVsModel(
object,
species = NULL,
ratio = FALSE,
log_scale = TRUE,
return_data = FALSE,
labels = TRUE,
show_unobserved = FALSE,
...
)
Arguments
object |
An object of class MizerParams or MizerSim. |
species |
The species to be included. Optional. By default all observed yields will be included. A vector of species names, or a numeric vector with the species indices, or a logical vector indicating for each species whether it is to be included (TRUE) or not. |
ratio |
Whether to plot model yield vs. observed yield (FALSE) or the ratio of model : observed yield (TRUE). Default is FALSE. |
log_scale |
Whether to plot on the log10 scale (TRUE) or not (FALSE). For the non-ratio plot this applies for both axes, for the ratio plot only the x-axis is on the log10 scale. Default is TRUE. |
return_data |
Whether to return the data frame for the plot (TRUE) or not (FALSE). Default is FALSE. |
labels |
Whether to show text labels for each species (TRUE) or not (FALSE). Default is TRUE. |
show_unobserved |
Whether to include also species for which no yield observation is available. If TRUE, these species will be shown as if their observed yield was equal to the model yield. |
... |
For |
Details
Before you can use this function you will need to have added a
yield_observed column to your model which gives the observed yield in
grams per year. For species for which you have no observed yield, you should set
the value in the yield_observed column to 0 or NA.
The total relative error is shown in the caption of the plot, calculated by
TRE = \sum_i|1-\rm{ratio_i}|
where
\rm{ratio_i} is the ratio of model yield / observed
yield for species i.
Value
A ggplot2 object with the plot of model yield by species compared
to observed yield. If return_data = TRUE, the data frame used to
create the plot is returned instead of the plot.
Examples
# create an example
params <- NS_params
species_params(params)$yield_observed <-
c(0.8, 61, 12, 35, 1.6, NA, 10, 7.6, 135, 60, 30, NA)
params <- calibrateYield(params)
# Plot with default options
plotYieldObservedVsModel(params)
# Plot including also species without observations
plotYieldObservedVsModel(params, show_unobserved = TRUE)
# Show the ratio instead
plotYieldObservedVsModel(params, ratio = TRUE)
Build the cumulative-distribution plot
Description
Internal worker shared by the plotCDF() methods. It integrates the spectra
data over size (optionally normalising), converts to a length axis if
requested, and either returns the data or draws the plot via
plotDataFrame().
Usage
plot_cdf(
plot_dat,
params,
power,
normalise,
log_x,
log_y,
wlim,
llim,
ylim,
highlight,
size_axis,
return_data
)
Arguments
plot_dat |
Spectra plotting data as produced for |
params |
A MizerParams object. |
power |
The power of weight that the abundance was multiplied by, used for the y-axis label. |
normalise |
If |
log_x, log_y |
Logical flags for log10 axes. |
wlim, llim |
Numeric vectors of length two giving the weight and length limits. |
ylim |
Numeric vector of length two giving the y-axis limits. |
highlight |
Name or vector of names of species to be highlighted. |
size_axis |
Either |
return_data |
If |
Value
A mizer_plot (ggplot2) object, or the data frame if
return_data = TRUE.
Build the diet-composition plot
Description
Internal worker shared by the plotDiet() methods. It melts the diet array
into a data frame, restricts to meaningful size ranges, converts to a length
axis if requested and draws a stacked-area plot of prey proportions.
Usage
plot_diet(
params,
n,
diet,
species,
log_x,
log_y,
wlim,
llim,
size_axis,
return_data
)
Arguments
params |
A MizerParams object. |
n |
Array of species abundances (species by size). |
diet |
Array of diet proportions (predator by size by prey). |
species |
The predator species to be plotted. |
log_x, log_y |
Logical flags for log10 axes. |
wlim, llim |
Numeric vectors of length two giving the weight and length limits. |
size_axis |
Either |
return_data |
If |
Value
A mizer_plot (ggplot2) object, or the plotting data frame if
return_data = TRUE.
Build the feeding-level plot
Description
Internal worker shared by the plotFeedingLevel() methods. It assembles the
plotting data, optionally adds the critical feeding level, restricts to each
species' size range, converts to a length axis if requested and draws the
plot.
Usage
plot_feeding_level(
params,
feed,
species,
highlight,
all.sizes,
include_critical,
log_x,
log_y,
wlim,
llim,
size_axis,
return_data
)
Arguments
params |
A MizerParams object. |
feed |
Array of feeding levels (species by size). |
species |
The species to be plotted. |
highlight |
Name or vector of names of species to be highlighted. |
all.sizes |
If |
include_critical |
Whether to also plot the critical feeding level. |
log_x, log_y |
Logical flags for log10 axes. |
wlim, llim |
Numeric vectors of length two giving the weight and length limits. |
size_axis |
Either |
return_data |
If |
Value
A mizer_plot (ggplot2) object, or the plotting data frame if
return_data = TRUE.
Build the growth-curves plot
Description
Internal worker shared by the plotGrowthCurves() methods. It computes the
modelled size at age, optionally adds a von Bertalanffy curve and observed
size-at-age data, and draws the plot.
Usage
plot_growth_curves(
params,
species,
max_age,
percentage,
species_panel,
highlight,
log_x,
log_y,
size_at_age,
return_data
)
Arguments
params |
A MizerParams object. |
species |
The species to be plotted. |
max_age |
The age up to which to plot the growth curve. |
percentage |
If |
species_panel |
If |
highlight |
Name or vector of names of species to be highlighted. |
log_x, log_y |
Logical flags for log10 axes. |
size_at_age |
Optional data frame of observed size-at-age data. |
return_data |
If |
Value
A mizer_plot (ggplot2) object, or the plotting data frame if
return_data = TRUE.
Validate the size-axis argument
Description
Validate the size-axis argument
Usage
plot_size_axis(size_axis = "w")
Arguments
size_axis |
Either |
Value
The matched size-axis string, either "w" or "l".
Assemble the tooltip variables for a size-axis plot
Description
Assemble the tooltip variables for a size-axis plot
Usage
plot_size_tooltip(size_axis, before = NULL, after = NULL)
Arguments
size_axis |
Either |
before, after |
Optional character vectors of variable names to place before and after the size variable in the tooltip. |
Value
A character vector of tooltip variable names.
Name of the x-variable for a given size axis
Description
Name of the x-variable for a given size axis
Usage
plot_size_x_var(size_axis)
Arguments
size_axis |
Either |
Value
"l" for a length axis, otherwise "w".
Axis label for a given size axis
Description
Axis label for a given size axis
Usage
plot_size_xlab(size_axis)
Arguments
size_axis |
Either |
Value
The axis label: "Length [cm]" for a length axis, otherwise
"Size [g]".
Choose the x-axis limits for a given size axis
Description
Choose the x-axis limits for a given size axis
Usage
plot_size_xlim(wlim, size_axis, llim = c(NA, NA))
Arguments
wlim |
Numeric vector of length two giving the weight limits. |
size_axis |
Either |
llim |
Numeric vector of length two giving the length limits. |
Value
llim for a length axis, otherwise wlim.
Build the size-spectrum plot
Description
Internal worker shared by the plotSpectra() methods. It assembles the
plotting data frame from the species and resource abundances, applies the
size and abundance limits, optionally converts to a length axis, and either
returns the data or draws the plot via plotDataFrame().
Usage
plot_spectra(
params,
n,
n_pp,
species,
wlim,
llim,
ylim,
power,
total,
resource,
background,
highlight,
log_x,
log_y,
size_axis,
return_data
)
Arguments
params |
A MizerParams object. |
n |
Array of species abundances (species by size). |
n_pp |
Vector of resource abundance. |
species |
The species to be plotted. |
wlim, llim |
Numeric vectors of length two giving the weight and length limits. |
ylim |
Numeric vector of length two giving the y-axis limits. |
power |
The abundance is multiplied by weight raised to this power. |
total |
Whether to include the total community abundance. |
resource |
Whether to include the resource spectrum. |
background |
Whether to include background species. |
highlight |
Name or vector of names of species to be highlighted. |
log_x, log_y |
Logical flags for log10 axes. |
size_axis |
Either |
return_data |
If |
Value
A mizer_plot (ggplot2) object, or the plotting data frame if
return_data = TRUE.
Description of the plotting functions
Description
Mizer provides a range of plotting functions for visualising the results of running a simulation, stored in a MizerSim object, or the initial state stored in a MizerParams object.
Details
The quickest way to make a standard plot is often to call plot() directly.
mizer provides plot() methods for MizerSim and MizerParams objects, and
also for the array classes returned by many summary and rate functions:
-
plot(<MizerSim>)produces a five-panel summary plot withplotFeedingLevel(),plotBiomass(),plotPredMort(),plotFMort()andplotSpectra(). -
plot(<MizerParams>)produces a three-panel summary plot withplotFeedingLevel(),plotPredMort()andplotSpectra()from the initial state. -
plot(<ArrayTimeBySpecies>)plots any time-by-species array, such as those returned bygetBiomass(),getSSB(),getYield()andgetN()on aMizerSim, as lines of value against time. -
plot(<ArraySpeciesBySize>)plots any species-by-size array, such as those returned bygetEncounter(),getFeedingLevel(),getPredMort(),getFMort()andgetSearchVolume(), as lines of value against body size. -
plot(<ArrayTimeBySpeciesBySize>)plots a time slice from a time-by-species-by-size array, such as those returned bygetFMort()orgetPredMort()on aMizerSim. -
plot(<ArrayResourceBySize>)plots a resource array, such as those returned bygetResourceMort(), against body size. -
plot(<ArrayTimeByResourceBySize>)plots a time slice from a time-by-resource array, such as that returned byNResource()on aMizerSim, against body size.
The same array objects can be passed to plotHover() to produce
hover-enabled plotly versions, for example plotHover(getBiomass(sim)) or
plotHover(getEncounter(params)). To add another compatible array to an
existing ggplot, use addPlot(). To compare two compatible mizer arrays
directly, use plot2(). To plot cumulative distributions over body size,
use plotCDF(). To visualise how spectra or rates change through time, use
animate() on a MizerSim or an
ArrayTimeBySpeciesBySize object.
The named plotting functions give more specialised control. This table shows the available named plotting functions.
| Plot | Description |
plotBiomass() | Plots the total biomass of each species through time. A time range to be plotted can be specified. The size range of the community can be specified in the same way as for getBiomass(). |
plotYield() | Plots the total yield of each species across all fishing gears against time. |
plotYieldGear() | Plots the total yield of each species by gear against time. |
plotSpectra() | Plots the abundance (biomass or numbers) spectra of each species and the background community. It is possible to specify a minimum size which is useful for truncating the plot. |
plotCDF() | Plots cumulative distributions of abundance or biomass over size. |
plotCDF2() | Compares cumulative distributions from two simulations or parameter objects in one plot. |
plotSpectra2() | Compares the spectra from two simulations or parameter objects in one plot. |
plotFeedingLevel() | Plots the feeding level of each species against size. |
plotPredMort() | Plots the predation mortality of each species against size. |
plotFMort() | Plots the total fishing mortality of each species against size. |
plotGrowthCurves() | Plots the size as a function of age. |
plotDiet() | Plots the diet composition at size for a given predator species. |
plotBiomassObservedVsModel() | Compares observed biomass with model biomass. |
plotYieldObservedVsModel() | Compares observed yield with model yield. |
animate() | Animates spectra or rate arrays through time. The older animateSpectra() name is retained as an alias.
|
The static plotting functions use ggplot2 and return a ggplot object. This
means that you can manipulate the plot further after its creation using the
ggplot grammar of graphics. The named high-level plot functions have plotly
counterparts, for example plotlyBiomass() or plotlySpectra(), for
interactive exploration. Generic and compositional plotting APIs, such as
plot(), plot2(), plotRelative() and addPlot(), do not have separate
plotly wrappers. Use plotHover() on the ggplot object they return.
While most plot functions take their data from a MizerSim object, some of those that make plots representing data at a single time can also take their data from the initial values in a MizerParams object.
Where plots show results for species, the line colour and line type for each
species are specified by the linecolour and linetype slots in
the MizerParams object. These were either taken from a default palette
hard-coded into emptyParams() or they were specified by the user
in the species parameters dataframe used to set up the MizerParams object.
The linecolour and linetype slots hold named vectors, named by
the species. They can be overwritten by the user at any time.
Most plots allow the user to select to show only a subset of species,
specified as a vector in the species argument to the plot function.
The ordering of the species in the legend is the same as the ordering in the species parameter data frame.
See Also
summary_functions, indicator_functions
Other plotting functions:
addPlot(),
animate(),
plot,
plot2(),
plotBiomass(),
plotCDF(),
plotCDF2(),
plotDiet(),
plotFMort(),
plotFeedingLevel(),
plotGrowthCurves(),
plotMizerParams,
plotMizerSim,
plotPredMort(),
plotRelative(),
plotSpectra(),
plotSpectra2(),
plotSpectraRelative(),
plotYield(),
plotYieldGear()
Examples
sim <- NS_sim
# Generic plot methods
plot(sim)
plot(getBiomass(sim), species = c("Cod", "Herring"))
plotHover(getBiomass(sim))
# Named plot functions
plotFeedingLevel(sim)
# Plotting only a subset of species
plotFeedingLevel(sim, species = c("Cod", "Herring"))
# Adding another compatible array to an existing plot
p <- plot(getBiomass(sim), species = "Cod")
addPlot(p, getBiomass(sim), species = "Herring", linetype = "dashed")
# Specifying new colours and linetypes for some species
sim@params@linetype["Cod"] <- "dashed"
sim@params@linecolour["Cod"] <- "red"
plotFeedingLevel(sim, species = c("Cod", "Herring"))
# Manipulating the plot
library(ggplot2)
p <- plotFeedingLevel(sim)
p <- p + geom_hline(aes(yintercept = 0.7))
p <- p + theme_bw()
p
Bin average of a power law over geometric bins
Description
Computes the exact average of the power law w^d over each bin
[w_j, w_{j+1}], i.e.
\overline{w^d}_j = \frac{1}{\Delta w_j}\int_{w_j}^{w_{j+1}} w^d\, dw.
Usage
power_law_bin_average(w, dw, d, w_max = Inf)
Arguments
w |
Numeric vector of left bin edges |
dw |
Numeric vector of bin widths |
d |
Single numeric exponent of the power law. |
w_max |
Optional upper cutoff. The power law is taken to be zero above
|
Details
The integral has a closed form, so the result is exact (not merely second order):
\overline{w^d}_j = \frac{w_{j+1}^{d+1} - w_j^{d+1}}{(d+1)\,\Delta w_j},
\quad d \neq -1,
\overline{w^d}_j = \frac{\ln(w_{j+1}/w_j)}{\Delta w_j},
\quad d = -1.
This is used by the bin-averaged (second-order) code paths that need the
average of a power-law rate over each bin, for example setExtMort() and
the resource capacity and rate in setResource(). The grid does not need to
be geometric; only the left bin edges w and the bin widths dw are used,
with w_{j+1} = w_j + \Delta w_j.
An optional upper cutoff w_max handles a knife-edge truncation of the power
law (for example the resource carrying capacity, which is \kappa
w^{-\lambda} below w_pp_cutoff and zero above it). The bin straddling the
cutoff then receives the partial bin-average — the power-law average over
the part of the bin below w_max, divided by the full bin width — and bins
entirely above the cutoff get zero.
Value
A numeric vector (same length as w) of bin averages of
w^d (truncated at w_max when supplied).
Power-law predation kernel
Description
This predation kernel is a power-law, with sigmoidal cut-offs at large and small predator/prey mass ratios.
Usage
power_law_pred_kernel(
ppmr,
kernel_exp,
kernel_l_l,
kernel_u_l,
kernel_l_r,
kernel_u_r
)
Arguments
ppmr |
A vector of predator/prey size ratios at which to evaluate the predation kernel. |
kernel_exp |
The exponent of the power law |
kernel_l_l |
The location of the left, rising sigmoid |
kernel_u_l |
The shape of the left, rising sigmoid |
kernel_l_r |
The location of the right, falling sigmoid |
kernel_u_r |
The shape of the right, falling sigmoid |
Details
The return value is calculated as
ppmr^kernel_exp /
(1 + (exp(kernel_l_l) / ppmr)^kernel_u_l) /
(1 + (ppmr / exp(kernel_l_r))^kernel_u_r)
The parameters need to be given as columns in the species parameter dataframe.
Value
A vector giving the value of the predation kernel at each of the
predator/prey mass ratios in the ppmr argument.
See Also
Other predation kernel:
box_pred_kernel(),
lognormal_pred_kernel(),
truncated_lognormal_pred_kernel()
Examples
params <- NS_params
# Set all required paramters before changing kernel type
species_params(params)["Cod", "kernel_exp"] <- -0.8
species_params(params)["Cod", "kernel_l_l"] <- 4.6
species_params(params)["Cod", "kernel_u_l"] <- 3
species_params(params)["Cod", "kernel_l_r"] <- 12.5
species_params(params)["Cod", "kernel_u_r"] <- 4.3
species_params(params)["Cod", "pred_kernel_type"] <- "power_law"
plot(w_full(params), getPredKernel(params)["Cod", 10, ], type="l", log="x")
Integrate spectra data into a cumulative distribution
Description
Multiplies the spectra density by the size-bin widths and forms the cumulative sum over size for each species, optionally normalising each curve to end at 1.
Usage
prepare_spectra_cdf_data(plot_dat, params, normalise = TRUE)
Arguments
plot_dat |
Spectra plotting data with a |
params |
A MizerParams object, used for the size-bin widths. |
normalise |
If |
Value
The plotting data with the value column replaced by its cumulative distribution over size.
Print mizer objects
Description
Mizer supplies print() methods for the array-like objects returned by many
rate and summary functions. These methods print a compact preview of the
underlying matrix, array or vector: a header reporting the value name,
dimensions and units, followed by the actual values, truncated to fit the
console when the array is large. Species are truncated to a leading
subset, sizes to an evenly log-spaced sample spanning the full size range
(because size grids are uniform in log-space, this shows small, medium and
large individuals rather than just the smallest), and time series to a
representative sample of time steps that always includes the first and
last. A trailing note reports how much was omitted. A three-dimensional
ArrayTimeBySpeciesBySize() object is previewed via its final time slice,
matching the default behaviour of plot() for that class.
Usage
## S3 method for class 'ArraySpeciesBySize'
print(x, ...)
## S3 method for class 'ArrayTimeBySpecies'
print(x, ...)
## S3 method for class 'ArrayTimeBySpeciesBySize'
print(x, ...)
## S3 method for class 'summary.ArraySpeciesBySize'
print(x, ...)
## S3 method for class 'summary.ArrayTimeBySpecies'
print(x, ...)
## S3 method for class 'summary.ArrayTimeBySpeciesBySize'
print(x, ...)
Arguments
x |
The object to print. |
... |
Further arguments. They are currently ignored by the mizer methods. |
Details
For full numeric access, use the object itself as an ordinary matrix, array
or vector, or convert it to a long data frame with as.data.frame().
Value
The printed object, invisibly.
See Also
summary(), as.data.frame(), plot(),
ArraySpeciesBySize(), ArrayTimeBySpecies(),
ArrayTimeBySpeciesBySize()
Examples
enc <- getEncounter(NS_params)
print(enc)
biomass <- getBiomass(NS_sim)
print(biomass)
Project size spectrum forward in time
Description
Runs the size spectrum model simulation. The function returns an object of type MizerSim that can then be explored with a range of summary_functions, indicator_functions and plotting_functions.
Usage
project(
object,
effort,
t_max = 100,
dt = 0.1,
t_save = 1,
t_start = 0,
initial_n,
initial_n_pp,
append = TRUE,
progress_bar = TRUE,
callback = NULL,
method = c("euler", "predictor_corrector", "tr_bdf2"),
...
)
Arguments
object |
Either a MizerParams object or a
MizerSim object (which contains a |
effort |
The effort of each fishing gear through time. See notes below. |
t_max |
The number of years the projection runs for. The default value is 100. When an effort array is supplied, this argument can be used to extend the simulation beyond the times specified in the effort array. See notes below. |
dt |
Time step of the solver. The default value is 0.1. When |
t_save |
The frequency with which the output is stored. The default value is 1. See notes below. |
t_start |
The the year of the start of the simulation. The simulation
will cover the period from |
initial_n |
|
initial_n_pp |
|
append |
A boolean that determines whether the new simulation results
are appended to the previous ones. Only relevant if |
progress_bar |
Either a boolean value to determine whether a progress bar should be shown in the console, or a shiny Progress object to implement a progress bar in a shiny app. |
callback |
A function to be called at each saved time step of the
simulation. The callback function is called with the |
method |
The numerical method to use for the consumer density update.
|
... |
Other arguments will be passed to rate functions. |
Value
An object of class MizerSim.
Advective flux scheme
The spatial discretisation of the growth (advection) term is controlled by
the flux entry of the second_order_w slot of the params object, not by
an argument to project(). With the default ("upwind") the first-order
upwind flux is used. Setting it to "van_leer" (for example with
second_order_w(params) <- TRUE) switches on a flux-limited (van Leer, TVD)
deferred correction that removes the leading numerical diffusion
\approx g\,w\,\log\beta of the upwind flux while keeping the density
update a tridiagonal solve and preserving positivity. The correction is most
useful on coarse logarithmic grids and pairs naturally with the second-order
time methods. Because it changes the discrete steady state, the choice lives
in the params object alongside the steady state rather than being a per-run
argument. See second_order_w().
Note
The effort argument specifies the level of fishing effort during the
simulation. If it is not supplied, the initial effort stored in the params
object is used. The effort can be specified in four different ways:
A single numeric value. This specifies the effort of all fishing gears which is constant through time (i.e. all the gears have the same constant effort).
A named vector whose names match with existing gear names. The values in the vector specify the constant fishing effort for those fishing gears, i.e. the effort is constant through time. The effort for gears that are not included in the effort vector is set to the default effort value, which is 1 in defaults edition 2 and later and 0 in earlier defaults editions. Missing (
NA) effort entries are replaced in the same way.A numerical vector which has the same length as the number of fishing gears. The values in the vector specify the constant fishing effort of each of the fishing gears, with the ordering assumed to be the same as in the MizerParams object.
A numerical array with dimensions time x gear. This specifies the fishing effort of each gear at each time step. The first dimension, time, must be named numerically and increasing. The second dimension of the array must be named and the names must correspond to the gear names in the
MizerParamsobject. The value for the effort for a particular time is used during the interval from that time to the next time in the array.
If effort is specified as an array then the smallest time in the array is
used as the initial time for the simulation. Otherwise the initial time is
set to the final time of the previous simulation if object is a
MizerSim object or to t_start otherwise.
When an effort array is provided, the t_max argument can be used to
extend the simulation beyond the last time specified in the effort array.
In this case, the effort values from the last time in the array will be
used for the extended period. The t_save argument can be used to specify
the frequency at which simulation results are saved. If t_save is not
supplied, the results will be saved at the times specified in the effort
array. If both t_max and t_save are provided with an effort array,
effort values will be interpolated (using step function) or extrapolated
(using the last known value) as needed for the new time points. The
t_start argument continues to be ignored when an effort array is supplied.
Note that if t_max or t_save are specified, the time grid for the
simulation is resampled based on t_save. This means that if the time
points in the effort array are irregular and do not align with the new
grid, those specific time points may be lost and the effort values at the
new grid points will be calculated via interpolation.
If the object argument is of class MizerSim then the initial
values for the simulation are taken from the final values in the
MizerSim object and the corresponding arguments to this function will
be ignored.
Examples
params <- NS_params
# With constant fishing effort for all gears for 20 time steps
sim <- project(params, t_max = 20, effort = 0.5)
# With constant fishing effort which is different for each gear
effort <- c(Industrial = 0, Pelagic = 1, Beam = 0.5, Otter = 0.5)
sim <- project(params, t_max = 20, effort = effort)
# With fishing effort that varies through time for each gear
gear_names <- c("Industrial", "Pelagic", "Beam", "Otter")
times <- seq(from = 1, to = 10, by = 1)
effort_array <- array(NA,
dim = c(length(times), length(gear_names)),
dimnames = list(time = times, gear = gear_names)
)
effort_array[, "Industrial"] <- 0.5
effort_array[, "Pelagic"] <- seq(from = 1, to = 2, length = length(times))
effort_array[, "Beam"] <- seq(from = 1, to = 0, length = length(times))
effort_array[, "Otter"] <- seq(from = 1, to = 0.5, length = length(times))
sim <- project(params, effort = effort_array)
# Extend a simulation beyond the effort array times
# Effort values from the final time are used for the extension
sim <- project(params, effort = effort_array, t_max = 15)
# Control save times with an effort array using t_save
sim <- project(params, effort = effort_array, t_save = 2)
Get density-dependent reproduction rate during projection
Description
S3 generic used by extension-aware projections to calculate the
density-dependent reproduction rate. The base method calls the selected
density-dependence function in params@rates_funcs$RDD.
Usage
projectRDD(params, rdi, species_params = params@species_params, t = 0, ...)
## S3 method for class 'MizerParams'
projectRDD(params, rdi, species_params = params@species_params, t = 0, ...)
Arguments
params |
A MizerParams object. |
rdi |
Vector of density-independent reproduction rates
|
species_params |
A species parameter dataframe. Must contain a column
|
t |
The time for which to do the calculation. |
... |
Unused |
Value
Vector of density-dependent reproduction rates.
Project to steady state
Description
Run the full dynamics, as in project(), but stop once the change has slowed
down sufficiently, in the sense that the distance between states at
successive time steps is less than tol. You determine how the distance is
calculated.
Usage
projectToSteady(
params,
effort = params@initial_effort,
distance_func = distanceSSLogN,
t_per = 1.5,
t_max = 100,
dt = 0.1,
tol = 0.1 * t_per,
return_sim = FALSE,
progress_bar = TRUE,
info_level = 3,
method = c("euler", "predictor_corrector", "tr_bdf2"),
...
)
Arguments
params |
A MizerParams object |
effort |
The fishing effort to be used throughout the simulation.
This is validated by |
distance_func |
A function that will be called after every |
t_per |
The simulation is broken up into shorter runs of |
t_max |
The maximum number of years to run the simulation. Default is 100. |
dt |
The time step to use in |
tol |
The simulation stops when the relative change in the egg
production RDI over |
return_sim |
If TRUE, the function returns the MizerSim object holding
the result of the simulation run, saved at intervals of |
progress_bar |
A shiny progress object to implement a progress bar in a shiny app. Default FALSE. |
info_level |
Controls the amount of information messages that are shown. Higher levels lead to more messages. |
method |
The numerical method to use for the consumer density update.
See |
... |
Further arguments will be passed on to your distance function. |
Value
If return_sim = FALSE, a MizerParams object with the initial
state replaced by the final state found by the steady-state search. If
return_sim = TRUE, a MizerSim object containing the intermediate states
saved every t_per years.
See Also
distanceSSLogN(), distanceMaxRelRDI()
Project values for first time step of Euler method
Description
This is an internal function used by the user-facing project() function.
It is of potential interest only to mizer extension authors.
Usage
project_n(
params,
r,
n,
dt,
a,
b,
c,
S,
idx,
w_min_idx_array_ref,
no_sp,
no_w,
flux_limiter = "none"
)
project_n_no_diffusion(
params,
r,
n,
dt,
a,
b,
S,
idx,
w_min_idx_array_ref,
no_sp,
no_w
)
Arguments
params |
A MizerParams object. |
r |
A list of rates as returned by |
n |
An array (species x size) with the number density at the current time step. |
dt |
Time step. |
a |
A matrix (species x size) used in the solver (transport term). |
b |
A matrix (species x size) used in the solver (diagonal term). |
c |
A matrix (species x size) used in the solver (transport term). |
S |
A matrix (species x size) used in the solver (source term). |
idx |
Index vector for size bins (excluding the first one). |
w_min_idx_array_ref |
Index vector for the start of the size spectrum for each species. |
no_sp |
Number of species. |
no_w |
Number of size bins. |
flux_limiter |
Name of the flux limiter used for a deferred high-order
correction of the upwind advective flux, or |
Details
The function calculates the abundance at the next time step using the McKendrick-von Foerster equation:
\frac{\partial N}{\partial t} + \frac{\partial}{\partial w} \left( g N - \frac{1}{2}\frac{\partial(D N)}{\partial w} \right) = -\mu N
which is solved using a semi-implicit upwind finite volume scheme.
Value
The updated abundance density matrix n.
See Also
Project values with a predictor-corrector method
Description
This is an experimental second-order time stepping variant of project_n().
It first predicts the new consumer densities with project_n(), optionally
recalculates rates from that prediction, and then applies a Crank-Nicolson
corrector using midpoint rates.
Usage
project_n_2(
params,
r,
n,
dt,
a,
b,
c,
S,
idx,
w_min_idx_array_ref,
no_sp,
no_w,
rates_fns = NULL,
n_pp = NULL,
n_other = NULL,
t = 0,
effort = NULL,
r_hat = NULL,
r_mid = NULL,
n_hat = NULL,
flux_limiter = "none",
...
)
Arguments
params |
A MizerParams object. |
r |
A list of rates as returned by |
n |
An array (species x size) with the number density at the current time step. |
dt |
Time step. |
a |
A matrix (species x size) used in the solver (transport term). |
b |
A matrix (species x size) used in the solver (diagonal term). |
c |
A matrix (species x size) used in the solver (transport term). |
S |
A matrix (species x size) used in the solver (source term). |
idx |
Index vector for size bins (excluding the first one). |
w_min_idx_array_ref |
Index vector for the start of the size spectrum for each species. |
no_sp |
Number of species. |
no_w |
Number of size bins. |
rates_fns |
Optional named list of rate functions, as used by
|
n_pp |
Resource abundance used when recalculating provisional rates. |
n_other |
Other ecosystem components used when recalculating provisional rates. |
t |
Current time. |
effort |
Fishing effort used when recalculating provisional rates. |
r_hat |
Optional provisional end-of-step rates. If supplied, these are used instead of recalculating them. |
r_mid |
Optional midpoint rates. If supplied, these are used directly in the Crank-Nicolson corrector. |
n_hat |
Optional provisional end-of-step densities. When supplied with a
flux limiter, the limiter is frozen at the midpoint field |
flux_limiter |
Name of the flux limiter used for a deferred high-order
correction of the upwind advective flux, or |
... |
Further arguments passed to the rate functions. |
Details
If the rate recalculation arguments are not supplied, the corrector uses the supplied rates as fixed rates. In that case the corrector is second order only for the frozen-rate transport problem, not for the full nonlinear mizer dynamics.
Value
The updated abundance density matrix n.
See Also
Project values with the TR-BDF2 method
Description
This is an L-stable, second-order time stepping variant of project_n(). It
takes one TR-BDF2 step, consisting of a trapezoidal (Crank-Nicolson) stage
over the first part of the time step followed by a second-order backward
differentiation (BDF2) stage over the remainder.
Usage
project_n_tr_bdf2(
params,
r,
n,
dt,
a,
b,
c,
S,
idx,
w_min_idx_array_ref,
no_sp,
no_w,
rates_fns = NULL,
n_pp = NULL,
n_other = NULL,
t = 0,
effort = NULL,
r_hat = NULL,
r_mid = NULL,
n_hat = NULL,
flux_limiter = "none",
...
)
Arguments
params |
A MizerParams object. |
r |
A list of rates as returned by |
n |
An array (species x size) with the number density at the current time step. |
dt |
Time step. |
a |
A matrix (species x size) used in the solver (transport term). |
b |
A matrix (species x size) used in the solver (diagonal term). |
c |
A matrix (species x size) used in the solver (transport term). |
S |
A matrix (species x size) used in the solver (source term). |
idx |
Index vector for size bins (excluding the first one). |
w_min_idx_array_ref |
Index vector for the start of the size spectrum for each species. |
no_sp |
Number of species. |
no_w |
Number of size bins. |
rates_fns |
Optional named list of rate functions, as used by
|
n_pp |
Resource abundance used when recalculating provisional rates. |
n_other |
Other ecosystem components used when recalculating provisional rates. |
t |
Current time. |
effort |
Fishing effort used when recalculating provisional rates. |
r_hat |
Optional provisional end-of-step rates. If supplied, these are used instead of recalculating them. |
r_mid |
Optional midpoint rates. If supplied, these are used directly to build the TR-BDF2 operator. |
n_hat |
Optional provisional end-of-step densities. When supplied with a
flux limiter, the limiter is frozen at the midpoint field |
flux_limiter |
Name of the flux limiter used for a deferred high-order
correction of the upwind advective flux, or |
... |
Further arguments passed to the rate functions. |
Details
The nonlinear rates are handled exactly as in project_n_2(): a provisional
Euler predictor gives end-of-step rates, which are averaged with the
start-of-step rates to obtain second-order-accurate midpoint rates r_mid.
Both TR-BDF2 stages then use this single frozen operator.
With the standard parameter \gamma = 2 - \sqrt 2 both stages share the
same implicit coefficient \alpha\,\Delta t with
\alpha = \gamma/2 = 1 - 1/\sqrt 2, so the operator
I - \alpha\,\Delta t\,L is assembled once with get_transport_coefs()
and each stage is a single tridiagonal solve with project_n_loop(), exactly
as in project_n() and project_n_2(). Unlike the Crank-Nicolson corrector
in project_n_2(), TR-BDF2 is L-stable and therefore damps the stiff modes
that cause Crank-Nicolson to oscillate at large time steps.
If the rate recalculation arguments are not supplied, the step uses the supplied rates as fixed rates. In that case the method is second order only for the frozen-rate transport problem, not for the full nonlinear mizer dynamics, but it remains L-stable.
Value
The updated abundance density matrix n.
See Also
Project abundances by a given number of time steps into the future
Description
This is an internal function used by the user-facing project() function.
It is of potential interest only to mizer extension authors.
Usage
project_simple(
params,
n,
n_pp,
n_other,
effort,
t,
dt,
steps,
resource_dynamics_fn,
other_dynamics_fns,
rates_fns,
method = c("euler", "predictor_corrector", "tr_bdf2"),
...
)
Arguments
params |
A MizerParams object. |
n |
An array (species x size) with the number density at start of simulation. |
n_pp |
A vector (size) with the resource number density at start of simulation. |
n_other |
A named list with the abundances of other components at start of simulation. |
effort |
The fishing effort to be used throughout the simulation. This must be a vector or list with one named entry per fishing gear. |
t |
Time at the start of the simulation. |
dt |
Size of time step. |
steps |
The number of time steps by which to project. |
resource_dynamics_fn |
The function for the resource dynamics. See Details. |
other_dynamics_fns |
List with the functions for the dynamics of the other components. See Details. |
rates_fns |
List with the functions for calculating the rates. See Details. |
method |
The numerical method to use for the consumer density update.
See |
... |
Other arguments that are passed on to the rate functions. |
Details
The function does not check its arguments because it is meant to be as fast
as possible to allow it to be used in a loop. For example, it is called in
project() once for every saved value. The function also does not save its
intermediate results but only returns the result at time t + dt * steps.
During this time it uses the constant fishing effort effort.
The functional arguments can be calculated from slots in the params object
with
resource_dynamics_fn <- get(params@resource_dynamics) other_dynamics_fns <- lapply(params@other_dynamics, get) rates_fns <- lapply(params@rates_funcs, get)
The reason the function does not do that itself is to shave 20 microseconds of its running time, which pays when the function is called hundreds of times in a row.
This function is also used in steady(). In between calls to
project_simple() the steady() function checks whether the values are
still changing significantly, so that it can stop when a steady state has
been approached. Mizer extension packages might have a similar need to run
a simulation repeatedly for short periods to run some other code in
between. Because this code may want to use the values of the rates from the
final update step, these too are included in the returned list.
Value
List with the final values of n, n_pp, and n_other, together
with rates, the rates calculated at the start of the final update step.
Does an installed extension register dispatch methods for its own class?
Description
An extension package participates in dispatch by registering S3 methods for
mizer generics keyed on its marker class (e.g. getEncounter.mizerMR). This
checks the package namespace's own S3 method registry for any method whose
class is the extension name or its sim variant. Because S3 method
registration does not require the S4 marker class to exist, this lets mizer
recognise a dispatching extension before creating its class, so extension
packages no longer have to define the marker class statically. Defining it
statically as contains = "MizerParams" would in fact prevent the package
from being chained with other extensions, since a sealed class cannot be
re-parented into the chain (see defineExtensionClasses()).
Usage
providesDispatchMethods(name)
Arguments
name |
The extension identifier (its marker class / package name). |
Value
TRUE if the loaded namespace name registers S3 methods for class
name or paste0(name, "Sim"), otherwise FALSE.
Record an extension and its version stamp on a mizer object
Description
Writes an entry for name into the object's @extensions slot, converting
the slot to the versioned list form. Existing entries (and their version
stamps) are preserved, keeping their position in the chain. A genuinely new
entry is prepended to the front of the chain so that it stays ordered
outermost-first, matching registerExtension(). The requirement is taken
from the existing entry if present, otherwise from the registered extension
chain.
Usage
recordExtension(params, name, version = NULL)
Arguments
params |
A |
name |
The extension identifier (its S4 marker class name). |
version |
Optional version string to stamp. If |
Details
Extension packages should call this instead of assigning to @extensions
directly. Pass version (typically packageVersion(name)) only when the
object has just been created or upgraded to conform to that version; leave it
NULL for ordinary modifications so the existing stamp is preserved.
Value
The params object with the updated @extensions slot.
See Also
"Creating a mizer extension package":
vignette("creating-extension-packages", package = "mizer")
Other extension tools:
NOther(),
clearExtensionChain(),
coerceToExtensionClass(),
getRegisteredExtensions(),
initialNOther<-(),
registerExtension(),
registerExtensions(),
setComponent(),
setRateFunction()
Objects exported from other packages
Description
These objects are imported from other packages. Follow the links below to see their documentation.
- reshape2
Register a single mizer extension for this R session
Description
Prepends one extension to the front of the active extension chain, giving it
the highest dispatch priority. Designed to be called from a package's
.onLoad hook so that the chain grows naturally in load order: the last
package loaded ends up outermost.
Usage
registerExtension(name, requirement = NA_character_, install = FALSE)
Arguments
name |
A syntactically valid R name identifying the extension (e.g.
|
requirement |
A version string, installation specification, or
|
install |
Logical. If |
Details
The call is idempotent: if the extension is already registered at any
position in the chain, the function returns silently without modifying the
chain. This makes it safe to call from devtools::load_all(), which
re-executes .onLoad.
Value
The updated extension chain, invisibly.
See Also
registerExtensions() for registering an explicit full chain.
"Using mizer extension packages":
vignette("using-extension-packages", package = "mizer").
"Creating a mizer extension package":
vignette("creating-extension-packages", package = "mizer")
Other extension tools:
NOther(),
clearExtensionChain(),
coerceToExtensionClass(),
getRegisteredExtensions(),
initialNOther<-(),
recordExtension(),
registerExtensions(),
setComponent(),
setRateFunction()
Register mizer extensions for this R session
Description
Registers an explicit full extension chain for the current R session. The
order of extensions is the S3 dispatch order, from outermost to innermost
extension. For example c(mizerExtB = "1.2.0", mizerExtA = "0.4.1")
dispatches to mizerExtB methods first, then mizerExtA methods, then base
mizer methods.
Usage
registerExtensions(extensions, install = FALSE)
Arguments
extensions |
A named character vector. Names are extension identifiers.
Values are version strings, installation specifications, or
|
install |
Logical. If |
Details
A session can handle objects whose extension chain is a suffix of the
registered maximal chain. For example, after registering
c(mizerExtB = "1.2.0", mizerExtA = "0.4.1"), objects using only
c(mizerExtA = "0.4.1") are also valid.
For extension packages that register themselves incrementally from .onLoad,
use registerExtension() instead.
Value
The active maximal extension chain, invisibly.
See Also
registerExtension() for the incremental per-package variant.
"Using mizer extension packages":
vignette("using-extension-packages", package = "mizer").
"Creating a mizer extension package":
vignette("creating-extension-packages", package = "mizer")
Other extension tools:
NOther(),
clearExtensionChain(),
coerceToExtensionClass(),
getRegisteredExtensions(),
initialNOther<-(),
recordExtension(),
registerExtension(),
setComponent(),
setRateFunction()
Symmetric relative difference between two values
Description
Symmetric relative difference between two values
Usage
relative_difference(first, second)
Arguments
first, second |
Numeric vectors to compare. |
Value
The symmetric relative difference
2 * (second - first) / (first + second).
Remove all background species
Description
Removes all species that have been marked as background species with
markBackground().
Usage
removeBackgroundSpecies(params)
Arguments
params |
A MizerParams object |
Details
This is just a shorthand for
removeSpecies(params, species_params(params)$is_background)
Value
A MizerParams object with background species removed
See Also
Examples
params <- markBackground(NS_params,
species = c("Sprat", "Sandeel", "N.pout"))
params <- removeBackgroundSpecies(params)
species_params(params)$species
Remove species
Description
This function simply removes all entries from the MizerParams object that refer to the selected species. It does not recalculate the steady state for the remaining species or retune their reproductive efficiency.
Usage
removeSpecies(params, species, ...)
Arguments
params |
A mizer params object for the original system. |
species |
The species to be removed. A vector of species names, or a numeric vector of species indices, or a logical vector indicating for each species whether it is to be removed (TRUE) or not. |
... |
Currently unused. |
Details
If a gear was targeting only the removed species, then this function will
NOT remove that gear. If you want to also remove that gear then you can do
that by calling setFishing().
Value
An object of type MizerParams
See Also
Examples
params <- NS_params
species_params(params)$species
params <- removeSpecies(params, c("Cod", "Haddock"))
species_params(params)$species
Rename gears
Description
Changes the names of gears in a MizerParams object. This involves for example changing the gear dimension names of selectivity and catchability arrays appropriately.
Usage
renameGear(params, replace, ...)
Arguments
params |
A mizer params object |
replace |
A named character vector, with new names as values, and old names as names. |
... |
Currently unused. |
Value
An object of type MizerParams
See Also
Examples
replace <- c(Industrial = "Trawl", Otter = "Beam_Trawl")
params <- renameGear(NS_params, replace)
gear_params(params)$gear
Rename species
Description
Changes the names of species in a MizerParams object. This involves for example changing the species dimension names of rate arrays appropriately.
Usage
renameSpecies(params, replace, ...)
Arguments
params |
A mizer params object |
replace |
A named character vector, with new names as values, and old names as names. |
... |
Currently unused. |
Value
An object of type MizerParams
See Also
Examples
replace <- c(Cod = "Kabeljau", Haddock = "Schellfisch")
params <- renameSpecies(NS_params, replace)
species_params(params)$species
Resolve a second_order_w value against the default scheme
Description
Internal helper that validates a second_order_w value against the default
first-order slot (flux = "upwind", bin_average = FALSE) and returns the
resulting named list. Used by the model constructors to work out the target
flux and bin_average entries before the rest of the model is built.
Usage
resolve_second_order_w(value)
Arguments
value |
The value to resolve, as accepted by |
Value
The resolved second_order_w list.
Keep resource abundance constant
Description
If you set your resource dynamics to use this function then the resource abundances are kept constant over time.
Usage
resource_constant(params, n_pp, ...)
Arguments
params |
A MizerParams object |
n_pp |
A vector of the resource abundance by size |
... |
Unused |
Details
To set your model to keep the resource constant over time you do
resource_dynamics(params) <- "resource_constant"
where you should replace params with the name of the variable holding your
MizerParams object.
Value
Vector containing the resource number density in each size class at the next timestep
See Also
Other resource dynamics functions:
resource_logistic(),
resource_semichemostat()
Examples
params <- NS_params
resource_dynamics(params) <- "resource_constant"
Project resource using logistic model
Description
If you set your resource dynamics to use this function then the time evolution of the resource spectrum is described by a logistic equation
\frac{\partial N_R(w,t)}{\partial t} = r_R(w) N_R(w)\Big[ 1 - \frac{N_R(w,t)}{c_R (w)} \Big] - \mu_R(w, t) N_R(w,t)
Usage
resource_logistic(
params,
n,
n_pp,
n_other,
rates,
t,
dt,
resource_rate,
resource_capacity,
...
)
balance_resource_logistic(params, resource_rate, resource_capacity)
Arguments
params |
A MizerParams object |
n |
A matrix of species abundances (species x size) |
n_pp |
A vector of the resource abundance by size |
n_other |
A list with the abundances of other components |
rates |
A list of rates as returned by |
t |
The current time |
dt |
Time step |
resource_rate |
Resource replenishment rate |
resource_capacity |
Resource carrying capacity |
... |
Unused |
Details
Here r_R(w) is the resource regeneration rate and c_R(w) is the
carrying capacity in the absence of predation. These parameters are changed
with setResource(). The mortality \mu_R(w, t) is
due to predation by consumers and is calculate with getResourceMort().
This function uses the analytic solution of the above equation to calculate
the resource abundance at time t + dt from all abundances and rates at time
t, keeping the mortality fixed during the timestep.
To set your model to use logistic dynamics for the resource you do
params <- setResource(params,
resource_dynamics = "resource_logistic",
resource_level = 0.5)
where you should replace params with the name of the variable holding your
MizerParams object. You can of course choose any value between 0 and 1 for
the resource level.
The balance_resource_logistic() function is called by setResource() to
determine the values of the resource parameters that are needed to make the
replenishment rate at each size equal the consumption rate at that size, as
calculated by getResourceMort(). It should be called with exactly one of
resource_rate or resource_capacity and returns a named list with values
for both. If resource_rate is supplied it must be at least as large as the
current mortality at each size. If resource_capacity is supplied it must be
not be less than the current resource abundance. Where it equals the
current resource abundance and there is positive consumption, it is nudged
upwards slightly to avoid division by zero.
Value
Vector containing the resource number density in each size class at the next timestep
See Also
Other resource dynamics functions:
resource_constant(),
resource_semichemostat()
Resource parameters
Description
The recommended way to change the resource dynamics parameters is to use
setResource(). The resource_params list contains values that are helpful
in setting up the actual size-dependent parameters with setResource(). If
you have specified a custom resource dynamics function that requires
additional parameters, then these should also be added to the
resource_params list.
Usage
resource_params(params)
resource_params(params) <- value
Arguments
params |
A MizerParams object |
value |
A named list of resource parameters. |
Details
The resource_params list will at least contain the slots kappa, lambda,
w_pp_cutoff and n.
The resource parameter n is the exponent for the power-law form for the
replenishment rate r_R(w):
r_R(w) = r_R\, w^{n-1}.
The resource parameter lambda (\lambda) is the exponent for the
power-law form for the carrying capacity c_R(w) and w_pp_cutoff is
its cutoff value:
c_R(w) = c_R w^{-\lambda}
for all w less than
w_pp_cutoff and zero for larger sizes.
The resource parameter kappa (\kappa) is the coefficient c_R of
the carrying capacity in the power law above, so
c_R(w) = \kappa\, w^{-\lambda}
for all w less than w_pp_cutoff and zero for larger sizes. Changing
kappa therefore rescales the carrying capacity. It has a second role in that
the same expression also set the initial resource abundance when the model was
created:
N_R(w) = \kappa\, w^{-\lambda}.
Unlike the carrying capacity, however, the initial resource abundance is
not updated when you subsequently change kappa (or call setResource()).
Assigning to resource_params only rebuilds the size-dependent resource rate
and capacity arrays from these scalars (leaving any arrays you have set
manually untouched). It does not balance the resource, i.e. it does not
adjust one of the rate or capacity to keep the resource at the steady state
where it replenishes at the rate at which it is consumed. This mirrors the
way the species parameters feed the species rates. If you want to preserve
the steady state after changing a resource scalar, call setResource() with
the appropriate argument (which balances by default).
Value
A named list of resource parameters.
See Also
Construct the background resource power-law spectrum
Description
Internal helper returning the auto-calculated resource power law
\kappa\, w^{-\lambda} on the full size grid, optionally truncated at an
upper cutoff w_max. When the bin_average entry of the model's
second_order_w slot is set, the exact bin average of the power law over
each bin is returned instead (with the bin straddling w_max getting the
partial average), so that the initial resource is the finite-volume cell
average of the background spectrum, consistent with the bin-integrated
encounter convolution that consumes it as a cell average. Otherwise the
left-edge point values are returned, byte-identical to previous mizer.
Usage
resource_power_law(params, kappa, lambda, w_max = Inf)
Arguments
params |
A MizerParams object whose |
kappa |
The coefficient |
lambda |
The exponent so the power law is |
w_max |
Optional upper cutoff. The power law is taken to be zero at and
above |
Details
This is used wherever the background resource spectrum \kappa
w^{-\lambda} is constructed from scratch: the initial resource abundance in
newMultispeciesParams() and the temporary prey spectra used to compute the
default gamma/f0 (get_gamma_default(), get_f0_default()) and the
consumer initial abundances (get_initial_n()). The bin-averaged resource
capacity and rate are produced directly by setResource().
Value
A numeric vector (same length as w_full) of the resource spectrum.
Project resource using semichemostat model
Description
If you set your resource dynamics to use this function then the time evolution of the resource spectrum is described by a semi-chemostat equation
\frac{\partial N_R(w,t)}{\partial t} = r_R(w) \Big[ c_R (w) - N_R(w,t) \Big] - \mu_R(w, t) N_R(w,t)
Usage
resource_semichemostat(
params,
n,
n_pp,
n_other,
rates,
t,
dt,
resource_rate,
resource_capacity,
...
)
balance_resource_semichemostat(params, resource_rate, resource_capacity)
Arguments
params |
A MizerParams object |
n |
A matrix of species abundances (species x size) |
n_pp |
A vector of the resource abundance by size |
n_other |
A list with the abundances of other components |
rates |
A list of rates as returned by |
t |
The current time |
dt |
Time step |
resource_rate |
Resource replenishment rate |
resource_capacity |
Resource carrying capacity |
... |
Unused |
Details
Here r_R(w) is the resource regeneration rate and c_R(w) is the
carrying capacity in the absence of predation. These parameters are changed
with setResource(). The mortality \mu_R(w, t) is
due to predation by consumers and is calculate with getResourceMort().
This function uses the analytic solution of the above equation to calculate
the resource abundance at time t + dt from all abundances and rates at time
t, keeping the mortality fixed during the timestep.
To set your model to use semichemostat dynamics for the resource you do
params <- setResource(params,
resource_dynamics = "resource_semichemostat",
resource_level = 0.5)
where you should replace params with the name of the variable holding your
MizerParams object. You can of course choose any value between 0 and 1 for
the resource level.
The balance_resource_semichemostat() function is called by setResource()
to determine the values of the resource parameters that are needed to make
the replenishment rate at each size equal the consumption rate at that size,
as calculated by getResourceMort(). It should be called with only one of
resource_rate or resource_capacity and returns a named list with values
for both. If resource_rate is supplied it must be positive wherever the
current resource mortality is positive. If resource_capacity is supplied it
must not be less than the current resource abundance. Where it equals the
current resource abundance and there is positive consumption, it is nudged
upwards slightly to avoid division by zero.
Value
Vector containing the resource number density in each size class at the next timestep
See Also
Other resource dynamics functions:
resource_constant(),
resource_logistic()
Run the registered extension upgrade methods on an object
Description
For each extension recorded in the object's @extensions slot (processed
innermost-first) whose installed package version is newer than the recorded
stamp (or whose stamp is missing), calls the extension's upgrade method if
one is registered, then records the installed version as the new stamp. The
core mizer upgrade is not run here; see upgrade.MizerParams().
Usage
runExtensionUpgrades(params)
Arguments
params |
A MizerParams object. |
Details
Extension upgrade methods are looked up with
getS3method("upgrade", name, optional = TRUE), must perform only their own
migration, must be idempotent, and must not call NextMethod().
Value
The object with extension migrations applied and stamps refreshed.
Save and restore mizer objects
Description
saveParams() saves a MizerParams object to a file. This can then be
restored with readParams(). saveSim() and readSim() provide the same
lifecycle for MizerSim objects.
Usage
saveParams(params, file)
readParams(file, install_extensions = FALSE)
saveSim(sim, file)
readSim(file, install_extensions = FALSE)
Arguments
params |
A MizerParams object |
file |
The name of the file or a connection where the object is saved to or read from. |
install_extensions |
Logical. Should |
sim |
A MizerSim object |
Details
While these functions ultimately use saveRDS() and readRDS(), they do
extra work to make the saved file more robust and more portable, so you
should always prefer them over calling saveRDS()/readRDS() directly on a
mizer object.
Value
saveParams() and saveSim() return NULL invisibly.
readParams() returns a MizerParams object. readSim() returns a MizerSim
object.
What saveParams() and saveSim() do beyond saveRDS()
They validate the object before writing it, so a corrupted or inconsistent object is caught at save time rather than when you next try to use it.
They strip any extension class and save the object as a plain base mizer object (recording which extension packages it needs in a slot). This means the file can be read back even in an R session where the extension packages that defined those S4 classes are not loaded, and it protects the file against future changes to those extension classes.
They check that the required extension packages are installed and stop with an informative error if they are not, so you do not save a file that you would be unable to read back.
They warn if the model relies on custom functions (custom rate, dynamics, selectivity or predation-kernel functions that are not provided by mizer or a registered extension package). Such functions are not stored in the file, so to share the model you also need to share an R script or R Markdown file defining them.
Before saving a model you may want to set its metadata with setMetadata().
What readParams() and readSim() do beyond readRDS()
They upgrade an object saved by an older version of mizer to the current structure (see
upgradeParams()), so that models saved years ago still load correctly.They re-register the extension packages that the model needs and, optionally, install any that are missing (see
install_extensions), before restoring the object's extension class.They coerce the object back to its extension class and revalidate it, reversing the class-stripping done at save time so you get back an object of the same class you saved.
See Also
"Using mizer extension packages":
vignette("using-extension-packages", package = "mizer")
Examples
# Save params to a temporary file and read them back
tmp <- tempfile(fileext = ".rds")
saveParams(NS_params, file = tmp)
params <- readParams(tmp)
identical(params, NS_params)
# Save and read back a simulation
tmp2 <- tempfile(fileext = ".rds")
saveSim(NS_sim, file = tmp2)
sim <- readSim(tmp2)
identical(sim, NS_sim)
Change scale of the model
Description
The abundances in mizer and some rates depend on the size of the area to which they refer. So they could be given per square meter or per square kilometre or for an entire study area or any other choice of yours. This function allows you to change the scale of the model by automatically changing the abundances and rates accordingly.
Usage
scaleModel(params, factor, ...)
Arguments
params |
A MizerParams object |
factor |
The factor by which the scale is multiplied |
... |
Additional arguments passed to the method. |
Details
If you rescale the model by a factor c then this function makes the
following rescalings in the params object:
The initial abundances are rescaled by
c.The search volume is rescaled by
1/c.The resource carrying capacity is rescaled by
cThe maximum reproduction rate
R_{max}is rescaled byc.
The effect of this is that the dynamics of the rescaled model are identical
to those of the unscaled model, in the sense that it does not matter whether
one first calls scaleModel() and then runs a simulation with
project() or whether one first runs a simulation and then rescales the
resulting abundances.
Note that if you use non-standard resource dynamics or other components then you may need to rescale additional parameters that appear in those dynamics.
In practice you will need to use some observations to set the scale for your
model. If you have biomass observations you can use calibrateBiomass(),
if you have yearly yields you can use calibrateYield().
Value
The rescaled MizerParams object
Rescale all rates in a mizer model
Description
Multiplies all rates in the model by a given factor. Rescaling all rates by
a factor f is equivalent to rescaling time by f: it speeds up
(or slows down) all dynamics without affecting the steady state of each
species, provided the resource spectrum is held at its steady-state value.
Usage
scaleRates(params, factor, ...)
Arguments
params |
A MizerParams object |
factor |
The positive factor by which all rates are multiplied. |
... |
Currently unused. |
Details
The following rates and their associated species parameters are rescaled:
Search volume (
search_volslot andgammaspecies parameter)Maximum intake rate (
intake_maxslot andhspecies parameter)Metabolic rate (
metabslot andks,kspecies parameters)External mortality (
mu_bslot andz0,z_ext,z0prespecies parameters)External encounter rate (
ext_encounterslot andE_extspecies parameter)External diffusion (
ext_diffusionslot andD_extspecies parameter)Catchability (
catchabilityslot andcatchabilitycolumn ingear_params)Maximum reproduction rate (
R_maxspecies parameter)Resource growth rate (
rr_ppslot)
Both the rate arrays stored in the MizerParams slots and the associated
species parameters in species_params and given_species_params are
rescaled, so that the parameters remain consistent with the rate arrays.
Value
The MizerParams object with all rates rescaled by factor.
See Also
Get or set the second_order_w flags
Description
Controls whether mizer uses numerical methods that are precise to second
order in \Delta w.
Usage
second_order_w(params)
second_order_w(params) <- value
Arguments
params |
A MizerParams object. |
value |
A single logical value ( |
Details
The slot is a named list with entries:
fluxThe advective-flux reconstruction scheme used in the numerical solver.
"upwind"is the first-order upwind scheme."van_leer"is the second-order scheme with the total-variation- diminishing van Leer limiter, which keeps abundances non-negative."centred"is the second-order scheme with the unlimited centred flux, which is genuinely second order even at extrema but is not monotonicity-preserving (it can produce small over/undershoots and is best used with some physical diffusion).bin_averageLogical. Controls whether bin-averaging is used for quantities that need it in order to be second-order precise in bin size. When
FALSE, point-sampling at the left bin edge is used.
When flux is "upwind" and bin_average is FALSE (the defaults),
mizer preserves the behaviour of previous mizer versions. Setting both to
their second-order values gives a consistently second-order model.
The setter accepts a single logical value (which sets both entries), a single
scheme name (which sets only flux), or a named vector to set individual
entries. The setter re-runs setParams() to rebuild precomputed arrays when
bin_average is changed.
Value
second_order_w(): A named list with entries flux (character) and
bin_average (logical).
second_order_w<-: A MizerParams object with the second_order_w
flags updated and, when bin_average is changed, all model parameters
recalculated via setParams().
Set Beverton-Holt reproduction without changing the steady state
Description
Takes a MizerParams object params with arbitrary density dependence in
reproduction and
returns a MizerParams object with Beverton-Holt density-dependence in such a
way that the energy invested into reproduction by the mature individuals
leads to the reproduction rate that is required to maintain the given egg
abundance. Hence if you have tuned your params object to describe a
particular steady state, then setting the Beverton-Holt density dependence
with this function will leave you with the exact same steady state. By
specifying one of the parameters erepro, R_max or reproduction_level
you pick the desired reproduction curve. More details of these parameters are
provided below.
Usage
setBevertonHolt(params, erepro, R_max, reproduction_level, ...)
Arguments
params |
A MizerParams object |
erepro |
Reproductive efficiency for each species. See details. |
R_max |
Maximum reproduction rate. See details. |
reproduction_level |
Sets |
... |
Unused
|
Details
With Beverton-Holt density dependence the relation between the energy
invested into reproduction and the number of eggs hatched is determined
by two parameters: the reproductive efficiency erepro and the maximum
reproduction rate R_max.
If no maximum is imposed on the reproduction rate
(R_{max} = \infty) then the resulting density-independent
reproduction rate R_{di} is proportional
to the total rate E_R at which energy is invested into reproduction,
R_{di} = \frac{\rm{erepro}}{2 w_{min}} E_R,
where the proportionality factor is given by the reproductive efficiency
erepro divided by the egg size w_min to convert energy to egg number and
divided by 2 to account for the two sexes.
Imposing a finite maximum reproduction rate R_{max} leads to a
non-linear relationship between energy invested and eggs hatched. This
density-dependent reproduction rate R_{dd} is given as
R_{dd} = R_{di}
\frac{R_{max}}{R_{di} + R_{max}}.
(All quantities in the above equations are species-specific but we dropped the species index for simplicity.)
The following plot illustrates the Beverton-Holt density dependence in the
reproduction rate for two different choices of parameters.
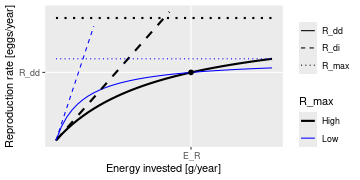
This plot shows that a given energy E_R invested into reproduction can
lead to the same reproduction rate R_{dd} with different choices
of the parameters R_max and erepro. R_max determines the asymptote of
the curve and erepro its initial slope. A higher R_max coupled with a
lower erepro (black curves) can give the same value as a lower R_max
coupled with a higher erepro (blue curves).
For the given initial state in the MizerParams object params one can
calculate the energy E_R that is invested into reproduction by the
mature individuals and the reproduction rate R_{dd} that is
required to keep the egg abundance constant. These two values determine the
location of the black dot in the above graph. You then only need one
parameter to select one curve from the family of Beverton-Holt curves going
through that point. This parameter can be erepro or R_max. Instead of
R_max you can alternatively specify the reproduction_level which is the
ratio between the density-dependent reproduction rate R_{dd} and
the maximal reproduction rate R_{max}.
If you do not provide a value for any of the reproduction parameter
arguments, then erepro will be set to the value it has in the current
species parameter data frame. If you do provide one of the reproduction
parameters, this can be either a vector with one value for each
species, or a named vector where the names determine which species are
affected, or a single unnamed value that is then used for all species. Any
species for which the given value is NA will remain unaffected.
The values for R_max must be larger than R_{dd} and can range
up to Inf. If a smaller value is requested a warning is issued and the
value is increased to the value required for a reproduction level of 0.99.
The values for the reproduction_level must be non-negative and
less than 1. The values for erepro must be large enough to allow the
required reproduction rate. If a smaller value is requested a warning is
issued and the value is increased to the smallest possible value. The values
for erepro should also be smaller than 1 to be physiologically sensible,
but this is not enforced by the function.
As can be seen in the graph above, choosing a lower value for R_max or a
higher value for erepro means that near the steady state the reproduction
will be less sensitive to a change in the energy invested into reproduction
and hence less sensitive to changes in the spawning stock biomass or its
energy income. As a result the species will also be less sensitive to
fishing, leading to a higher F_MSY.
Value
A MizerParams object
Examples
params <- NS_params
species_params(params)$erepro
# Attempting to set the same erepro for all species
params <- setBevertonHolt(params, erepro = 0.1)
t(species_params(params)[, c("erepro", "R_max")])
# Setting erepro for some species
params <- setBevertonHolt(params, erepro = c("Gurnard" = 0.6, "Plaice" = 0.95))
t(species_params(params)[, c("erepro", "R_max")])
# Setting R_max
R_max <- 1e17 * species_params(params)$w_max^-1
params <- setBevertonHolt(NS_params, R_max = R_max)
t(species_params(params)[, c("erepro", "R_max")])
# Setting reproduction_level
params <- setBevertonHolt(params, reproduction_level = 0.3)
t(species_params(params)[, c("erepro", "R_max")])
Set line colours and line types to be used in mizer plots
Description
Used for setting the colour and type of lines representing "Total",
"Resource", "Fishing", "Background", "External" and possibly other categories
in plots.
Usage
setColours(params, colours)
getColours(params)
setLinetypes(params, linetypes)
getLinetypes(params)
Arguments
params |
A MizerParams object |
colours |
A named list or named vector of line colours. |
linetypes |
A named list or named vector of linetypes. |
Details
Colours for names that already had a colour set for them will be overwritten by the colour you specify. Colours for names that did not yet have a colour will be appended to the list of colours.
If a name coincides with the name of a species, the linecolour (for
setColours()) or linetype (for setLinetypes()) entry for that species
in species_params and given_species_params is updated as well, so that
the choice persists with the species. Alternatively you can set the
linecolour and linetype variables in the species parameter data frame
directly, see the example below.
You can use the same colours in your own ggplot2 plots by adding
scale_colour_manual(values = getColours(params)) to your plot. Similarly
you can use the linetypes with
scale_linetype_manual(values = getLinetypes(params)).
Value
setColours: The MizerParams object with updated line colours
getColours(): A named vector of colours
setLinetypes(): The MizerParams object with updated linetypes
getLinetypes(): A named vector of linetypes
Examples
params <- setColours(NS_params, list("Resource" = "red","Total" = "#0000ff"))
params <- setLinetypes(NS_params, list("Total" = "dotted"))
# Set colours and linetypes for species, either via setColours()/
# setLinetypes() or directly via the species parameter data frame
params <- setColours(params, list("Cod" = "black"))
species_params(params)["Cod", "linetype"] <- "dashed"
plotSpectra(params, total = TRUE)
getColours(params)
getLinetypes(params)
Add a dynamical ecosystem component
Description
By default, mizer models any number of size-resolved consumer species and a single size-resolved resource spectrum. Your model may require additional components, like for example detritus or carrion or multiple resources or .... This function allows you to set up such components.
Usage
setComponent(
params,
component,
initial_value,
dynamics_fun,
encounter_fun,
mort_fun,
component_params,
colour = "grey",
linetype = "solid"
)
removeComponent(params, component)
getComponent(params, component)
Arguments
params |
A MizerParams object |
component |
Name of the component of interest. If missing, a list of all components will be returned. |
initial_value |
Initial value of the component |
dynamics_fun |
Name of function to calculate value at the next time step |
encounter_fun |
Name of function to calculate contribution to encounter rate. Optional. |
mort_fun |
Name of function to calculate contribution to the mortality rate. Optional. |
component_params |
Object holding the parameters needed by the component functions. This could for example be a named list of parameters. Optional. |
colour |
Line colour to use for the component in plots. Defaults to
|
linetype |
Line type to use for the component in plots. Defaults to
|
Details
The component can be a number, a vector, an array, a list, or any other data structure you like.
If you set a component with a new name, the new component will be added
to the existing components. If you set a component with an existing name,
the initial_value and dynamics_fun are overwritten, while the optional
encounter_fun, mort_fun and component_params are only changed if the
corresponding arguments are supplied. You can remove a component with
removeComponent().
Value
The updated MizerParams object
For getComponent: A list with the entries initial_value, dynamics_fun,
encounter_fun, mort_fun, component_params for the requested
component. If the requested component does not exist, NULL is returned.
If no component argument is given, then a list of lists for all
components is returned.
See Also
"Extending mizer":
vignette("extending-mizer", package = "mizer")
Other extension tools:
NOther(),
clearExtensionChain(),
coerceToExtensionClass(),
getRegisteredExtensions(),
initialNOther<-(),
recordExtension(),
registerExtension(),
registerExtensions(),
setRateFunction()
Set external diffusion rate
Description
You will usually not need to call this function directly. Instead change
the D_ext and n species parameters with
given_species_params(params) <- and let mizer recalculate the external
diffusion rate for you. Call setExtDiffusion() directly only if you want
to impose a different functional form for the size dependence of the
external diffusion rate. See vignette("cheatsheet-changing-parameters")
for a full explanation of when to reach for which level of the model.
Usage
setExtDiffusion(params, ext_diffusion = NULL, reset = FALSE, ...)
ext_diffusion(params)
ext_diffusion(params) <- value
Arguments
params |
MizerParams |
ext_diffusion |
Optional. An array (species x size) holding the
external diffusion rate. If not supplied, a default is calculated from the
|
reset |
If set to TRUE, then the external diffusion rate will be reset to the value calculated from the species parameters, even if it was previously overwritten with a custom value. If set to FALSE (default) then a recalculation from the species parameters will take place only if no custom value has been set. |
... |
Unused |
value |
ext_diffusion |
Value
setExtDiffusion(): A MizerParams object with updated external
diffusion rate.
ext_diffusion(): An ArraySpeciesBySize object (species x size)
with the external diffusion rate.
Setting external diffusion rate
The external diffusion rate allows you to impose additional diffusion beyond the predation-driven diffusion that can be internally modelled by mizer.
The ext_diffusion argument allows you to specify a diffusion rate that
depends on species and body size.
If the ext_diffusion argument is not supplied, then the external diffusion
rate is calculated as a power law:
D_{ext.i}(w) = D_{ext.i}\, w^{n_i+1}.
The coefficient D_{ext.i} is taken from the D_ext column of the
species parameter data frame, which defaults to 0. The exponent
n_i + 1 uses the n column of the species parameter data frame.
If the ext_diffusion slot has a comment and reset = FALSE, then a
recalculation from the species parameters is suppressed and a message is
issued if the recalculated values would differ from the stored ones.
See Also
Other functions for setting parameters:
gear_params(),
setExtEncounter(),
setExtMort(),
setFishing(),
setInteraction(),
setMaxIntakeRate(),
setMetabolicRate(),
setParams(),
setPredKernel(),
setReproduction(),
setSearchVolume(),
species_params(),
use_predation_diffusion()
Set external encounter rate
Description
You will usually not need to call this function directly. Instead change
the E_ext and n species parameters with
given_species_params(params) <- and let mizer recalculate the external
encounter rate for you. Call setExtEncounter() directly only if you want
to impose a different functional form for the size dependence of the
external encounter rate. See vignette("cheatsheet-changing-parameters")
for a full explanation of when to reach for which level of the model.
Usage
setExtEncounter(params, ext_encounter = NULL, reset = FALSE, ...)
getExtEncounter(params)
ext_encounter(params)
ext_encounter(params) <- value
Arguments
params |
MizerParams |
ext_encounter |
Optional. An array (species x size) holding the external
encounter rate. If not supplied, a default is calculated from the |
reset |
If set to TRUE, then the external encounter rate will be reset to the value calculated from the species parameters, even if it was previously overwritten with a custom value. If set to FALSE (default) then a recalculation from the species parameters will take place only if no custom value has been set. |
... |
Unused |
value |
ext_encounter |
Value
setExtEncounter(): A MizerParams object with updated external encounter
rate.
getExtEncounter() or equivalently ext_encounter(): A
ArraySpeciesBySize object (species x size) with the external encounter rate.
Setting external encounter rate
The external encounter rate is the rate at which a predator encounters food that is not explicitly modelled. It is a rate with units mass/year.
The ext_encounter argument allows you to specify an external encounter rate
that depends on species and body size. You can see an example of this in
the Examples section of the help page for setExtEncounter().
If the ext_encounter argument is not supplied, then the external encounter
rate is calculated as a power law:
E_{ext.i}(w) = E_{ext.i}\, w^{n_i}.
The coefficient E_{ext.i} is taken from the E_ext column of the
species parameter data frame, which defaults to 0. The exponent n_i is
taken from the n column of the species parameter data frame.
If the ext_encounter slot has a comment and reset = FALSE, then a
recalculation from the species parameters is suppressed and a message is
issued if the recalculated values would differ from the stored ones.
See Also
Other functions for setting parameters:
gear_params(),
setExtDiffusion(),
setExtMort(),
setFishing(),
setInteraction(),
setMaxIntakeRate(),
setMetabolicRate(),
setParams(),
setPredKernel(),
setReproduction(),
setSearchVolume(),
species_params(),
use_predation_diffusion()
Examples
params <- newMultispeciesParams(NS_species_params)
#### Setting allometric encounter rate #######################
# Set coefficient for each species. Here we choose 0.1 for each species
encounter_pre <- rep(0.1, nrow(species_params(params)))
# Multiply by power of size with exponent, here chosen to be 3/4
# The outer() function makes it an array species x size
allo_encounter <- outer(encounter_pre, w(params)^(3/4))
# Change the external encounter rate in the params object
ext_encounter(params) <- allo_encounter
Set external mortality rate
Description
You will usually not need to call this function directly. Instead change
the z0, z_ext and d species parameters with
given_species_params(params) <- and let mizer recalculate the external
mortality rate for you. Call setExtMort() directly only if you want to
impose a different functional form for the size dependence of the external
mortality. See vignette("cheatsheet-changing-parameters") for a full
explanation of when to reach for which level of the model.
Usage
setExtMort(
params,
ext_mort = NULL,
z0pre = 0.6,
z0exp = params@resource_params$n - 1,
reset = FALSE,
z0 = deprecated(),
...
)
getExtMort(params)
ext_mort(params)
ext_mort(params) <- value
Arguments
params |
MizerParams |
ext_mort |
Optional. An array (species x size) holding the external mortality rate. If not supplied, a default is set as described in the section "Setting external mortality rate". |
z0pre |
If |
z0exp |
If |
reset |
If set to TRUE, then the external mortality rate will be reset to the value calculated from the species parameters, even if it was previously overwritten with a custom value. If set to FALSE (default) then a recalculation from the species parameters will take place only if no custom value has been set. |
z0 |
|
... |
Unused |
value |
ext_mort |
Value
setExtMort(): A MizerParams object with updated external mortality
rate.
getExtMort() or equivalently ext_mort(): An ArraySpeciesBySize
object (species x size) with the external mortality.
Setting external mortality rate
The external mortality is all the mortality that is not due to fishing or predation by predators included in the model. The external mortality could be due to predation by predators that are not explicitly included in the model (e.g. mammals or seabirds) or due to other causes like illness. It is a rate with units 1/year.
The ext_mort argument allows you to specify an external mortality rate
that depends on species and body size. You can see an example of this in
the Examples section of the help page for setExtMort().
If the ext_mort argument is not supplied, then the external mortality is
taken from the species parameters as
\mu_{ext.i}(w) = z_{0.i} + z_{ext.i} w^{d_i}.
The value of the constant z_0 for each species is taken from the z0
column of the species parameter data frame, if that column exists.
Otherwise it is calculated as
z_{0.i} = {\tt z0pre}_i\, w_{inf}^{\tt z0exp}.
Missing values of z_ext are set to 0 and missing values of d are set to
n - 1.
By default the power law is evaluated at the left bin edges w_j
(point sampling). If the bin_average entry of the second_order_w slot is
TRUE (see second_order_w()), then the z_{ext} w^d term is instead
replaced by its exact average over each bin [w_j, w_{j+1}],
\frac{z_{ext}}{\Delta w_j}\int_{w_j}^{w_{j+1}} w^d\, dw
= z_{ext}\,\frac{w_{j+1}^{d+1} - w_j^{d+1}}{(d+1)\,\Delta w_j},
(with the limiting form z_{ext}\ln(w_{j+1}/w_j)/\Delta w_j when
d = -1). This is the consistent choice in the finite-volume scheme,
where the external mortality multiplies the bin-averaged abundance. The
bin-averaging is applied only to the auto-calculated power-law default; a
user-supplied ext_mort array is left untouched.
See Also
Other functions for setting parameters:
gear_params(),
setExtDiffusion(),
setExtEncounter(),
setFishing(),
setInteraction(),
setMaxIntakeRate(),
setMetabolicRate(),
setParams(),
setPredKernel(),
setReproduction(),
setSearchVolume(),
species_params(),
use_predation_diffusion()
Examples
params <- newMultispeciesParams(NS_species_params)
#### Setting allometric death rate #######################
# Set coefficient for each species. Here we choose 0.1 for each species
z0pre <- rep(0.1, nrow(species_params(params)))
# Multiply by power of size with exponent, here chosen to be -1/4
# The outer() function makes it an array species x size
allo_mort <- outer(z0pre, w(params)^(-1/4))
# Change the external mortality rate in the params object
ext_mort(params) <- allo_mort
Set fishing parameters
Description
Set fishing parameters
Usage
setFishing(
params,
selectivity = NULL,
catchability = NULL,
reset = FALSE,
initial_effort = NULL,
...
)
getCatchability(params)
catchability(params)
catchability(params) <- value
getSelectivity(params)
selectivity(params)
selectivity(params) <- value
getInitialEffort(params)
Arguments
params |
A MizerParams object |
selectivity |
Optional. An array (gear x species x size) that holds the
selectivity of each gear for species and size, |
catchability |
Optional. An array (gear x species) that holds the catchability of
each species by each gear, |
reset |
If set to TRUE, then both |
initial_effort |
Optional. A number or a named numeric vector specifying the fishing effort. If a number, the same effort is used for all gears. If a vector, must be named by gear. |
... |
Unused |
value |
The array to assign |
Value
setFishing(): A MizerParams object with updated fishing
parameters.
getCatchability() or equivalently catchability(): An array (gear
x species) that holds the catchability of each species by each gear,
Q_{g,i}. The names of the dimensions are "gear, "sp".
getSelectivity() or equivalently selectivity(): An array (gear x
species x size) that holds the selectivity of each gear for species and
size, S_{g,i,w}. The names of the dimensions are "gear, "sp", "w".
getInitialEffort() or equivalently initial_effort(): A named
vector with the initial fishing effort for each gear.
Setting fishing
Gears
In mizer, fishing mortality is imposed on species by fishing gears. The
total per-capita fishing mortality (1/year) is obtained by summing over the
mortality from all gears,
\mu_{f.i}(w) = \sum_g F_{g,i}(w),
where the fishing mortality F_{g,i}(w) imposed by gear g on
species i at size w is calculated as:
F_{g,i}(w) = S_{g,i}(w) Q_{g,i} E_{g},
where S is the selectivity by species, gear and size, Q is the
catchability by species and gear and E is the fishing effort by gear.
Selectivity
The selectivity at size of each gear for each species is saved as a three
dimensional array (gear x species x size). Each entry has a range between 0
(that gear is not selecting that species at that size) to 1 (that gear is
selecting all individuals of that species of that size). This three
dimensional array can be specified explicitly via the selectivity
argument, but usually mizer calculates it from the gear_params slot of
the MizerParams object.
To allow the calculation of the selectivity array, the gear_params slot
must be a data frame with one row for each gear-species combination. So if
for example a gear can select three species, then that gear contributes three
rows to the gear_params data frame, one for each species it can select. The
data frame must have columns gear, holding the name of the gear, species,
holding the name of the species, and sel_func, holding the name of the
function that calculates the selectivity curve. Some selectivity functions
are included in the package: knife_edge(), sigmoid_length(),
double_sigmoid_length(), and sigmoid_weight().
Users are able to write their own size-based selectivity function. The first
argument to the function must be w and the function must return a vector of
the selectivity (between 0 and 1) at size.
Each selectivity function may have parameters. Values for these
parameters must be included as columns in the gear parameters data.frame.
The names of the columns must exactly match the names of the corresponding
arguments of the selectivity function. For example, the default selectivity
function is knife_edge() that a has sudden change of selectivity from 0 to 1
at a certain size. In its help page you can see that the knife_edge()
function has arguments w and knife_edge_size. The first argument, w, is
size (the function calculates selectivity at size). All selectivity functions
must have w as the first argument. The values for the other arguments must
be found in the gear parameters data.frame. So for the knife_edge()
function there should be a knife_edge_size column. Because knife_edge()
is the default selectivity function, the knife_edge_size argument has a
default value = w_mat.
The most commonly-used selectivity function is sigmoid_length(). It has a
smooth transition from 0 to 1 at a certain size. The sigmoid_length()
function has the two parameters l50 and l25 that are the lengths in cm at
which 50% or 25% of the fish are selected by the gear. If you choose this
selectivity function then the l50 and l25 columns must be included in the
gear parameters data.frame.
In case each species is only selected by one gear, the columns of the
gear_params data frame can alternatively be provided as columns of the
species_params data frame, if this is more convenient for the user to set
up. Mizer will then copy these columns over to create the gear_params data
frame when it creates the MizerParams object. However changing these columns
in the species parameter data frame later will not update the gear_params
data frame.
Catchability
Catchability is used as an additional factor to make the link between gear selectivity, fishing effort and fishing mortality. For example, it can be set so that an effort of 1 gives a desired fishing mortality. In this way effort can then be specified relative to a 'base effort', e.g. the effort in a particular year.
Catchability is stored as a two dimensional array (gear x species). This can
either be provided explicitly via the catchability argument, or the
information can be provided via a catchability column in the gear_params
data frame.
In the case where each species is selected by only a single gear, the
catchability column can also be provided in the species_params data
frame. Mizer will then copy this over to the gear_params data frame when
the MizerParams object is created.
Effort
The initial fishing effort is stored in the MizerParams object. If it is
not supplied, it is set to zero. The initial effort can be overruled when
the simulation is run with project(), where it is also possible to specify
an effort that varies through time.
See Also
Other functions for setting parameters:
gear_params(),
setExtDiffusion(),
setExtEncounter(),
setExtMort(),
setInteraction(),
setMaxIntakeRate(),
setMetabolicRate(),
setParams(),
setPredKernel(),
setReproduction(),
setSearchVolume(),
species_params(),
use_predation_diffusion()
Examples
# Halve the initial fishing effort for all gears
params <- setFishing(NS_params, initial_effort = 0.5)
getInitialEffort(params)
str(getCatchability(NS_params))
str(getSelectivity(NS_params))
str(getInitialEffort(NS_params))
Set initial values to values from a simulation
Description
This function is deprecated. Use getParams(), initialParams(), or
finalParams() instead. These functions return a MizerParams object
with the ecosystem state extracted from a simulation.
Usage
setInitialValues(params, sim, time_range, geometric_mean = FALSE, ...)
Arguments
params |
A |
sim |
A |
time_range |
The time range to average the abundances over. Can be a vector of values, a vector of min and max time, or a single value. Only the range of times is relevant, i.e., all times between the smallest and largest will be selected. Default is the final time step. |
geometric_mean |
|
... |
Additional arguments passed to the method. |
Details
Value
The params object with updated initial values and initial effort.
Examples
params <- NS_params
sim <- project(params, t_max = 20, effort = 0.5)
params <- setInitialValues(params, sim)
Set species interaction matrix
Description
Set species interaction matrix
Usage
setInteraction(params, interaction = NULL, ...)
interaction_matrix(params)
interaction_matrix(params) <- value
Arguments
params |
MizerParams object |
interaction |
Optional interaction matrix of the species (predator species x prey species). By default all entries are 1. See "Setting interaction matrix" section below. |
... |
Unused |
value |
An interaction matrix |
Value
setInteraction: A MizerParams object with updated interaction
matrix
interaction_matrix(): The interaction matrix (predator species x
prey species)
Setting interaction matrix
You do not need to specify an interaction matrix. If you do not, then the predator-prey interactions are purely determined by the size of predator and prey and totally independent of the species of predator and prey.
The interaction matrix \theta_{ij} modifies the interaction of each
pair of species in the model. This can be used for example to allow for
different spatial overlap among the species.
The values in the interaction matrix are used to scale the encountered food
and predation mortality (see on the website the section on predator-prey encounter rate
and on predation mortality).
The first index refers to the predator species and the second to the prey
species.
The interaction matrix is used when calculating the food encounter rate in
getEncounter() and the predation mortality rate in getPredMort(). Its
entries are dimensionless numbers. If all the values in the interaction
matrix are equal then predator-prey interactions are determined entirely by
size-preference.
This function checks that the supplied interaction matrix is valid and then
stores it in the interaction slot of the params object.
The order of the columns and rows of the interaction argument should be
the same as the order in the species params data frame in the params
object. If you supply a named array then the function will check the order
and message if it is different before ignoring the supplied dimnames. If
you supply only column names then these are also used as the row names. One
way of creating your own interaction
matrix is to enter the data using a spreadsheet program and saving it as a
.csv file. The data can then be read into R using the command read.csv().
The interaction of the species with the resource are set via a column
interaction_resource in the species_params data frame. By default this
column is set to all 1s.
See Also
Other functions for setting parameters:
gear_params(),
setExtDiffusion(),
setExtEncounter(),
setExtMort(),
setFishing(),
setMaxIntakeRate(),
setMetabolicRate(),
setParams(),
setPredKernel(),
setReproduction(),
setSearchVolume(),
species_params(),
use_predation_diffusion()
Examples
params <- newTraitParams(no_sp = 3)
inter <- getInteraction(params)
inter[1, 2:3] <- 0
params <- setInteraction(params, interaction = inter)
getInteraction(params)
Set maximum intake rate
Description
You will usually not need to call this function directly. Instead change
the h and n species parameters with given_species_params(params) <-
and let mizer recalculate the maximum intake rate for you. Call
setMaxIntakeRate() directly only if you want to impose a different
functional form for the size dependence of the intake rate. See
vignette("cheatsheet-changing-parameters") for a full explanation of when
to reach for which level of the model.
Usage
setMaxIntakeRate(params, intake_max = NULL, reset = FALSE, ...)
getMaxIntakeRate(params)
intake_max(params)
intake_max(params) <- value
Arguments
params |
MizerParams |
intake_max |
Optional. An array (species x size) holding the maximum intake rate for each species at size. If not supplied, a default is set as described in the section "Setting maximum intake rate". |
reset |
If set to TRUE, then the intake rate will be reset to the value calculated from the species parameters, even if it was previously overwritten with a custom value. If set to FALSE (default) then a recalculation from the species parameters will take place only if no custom value has been set. |
... |
Unused |
value |
intake_max |
Value
setMaxIntakeRate(): A MizerParams object with updated maximum
intake rate.
getMaxIntakeRate() or equivalently intake_max(): A
ArraySpeciesBySize object (species x size) with the maximum intake rate.
Setting maximum intake rate
The maximum intake rate h_i(w) of an individual of species i and
weight w determines the feeding level, calculated with
getFeedingLevel(). It is measured in grams/year.
If the intake_max argument is not supplied, then the maximum intake
rate is set to
h_i(w) = h_i w^{n_i}.
The values of h_i (the maximum intake rate of an individual of size 1
gram) and n_i (the allometric exponent for the intake rate) are taken
from the h and n columns in the species parameter dataframe. If
the h column is not supplied in the species parameter dataframe, it is
calculated by the get_h_default() function. If the n column is not
supplied, a default of n_i = 3/4 is used.
If h_i is set to Inf, fish of species i will consume all encountered
food.
If the intake_max slot has a comment and reset = FALSE, then a
recalculation from the species parameters is suppressed and a message is
issued if the recalculated values would differ from the stored ones.
See Also
Other functions for setting parameters:
gear_params(),
setExtDiffusion(),
setExtEncounter(),
setExtMort(),
setFishing(),
setInteraction(),
setMetabolicRate(),
setParams(),
setPredKernel(),
setReproduction(),
setSearchVolume(),
species_params(),
use_predation_diffusion()
Examples
# Inspect the current maximum intake rate
getMaxIntakeRate(NS_params)["Cod", 1:5]
# Increase intake rate for Cod by 50%
intake_max <- getMaxIntakeRate(NS_params)
intake_max["Cod", ] <- intake_max["Cod", ] * 1.5
params <- setMaxIntakeRate(NS_params, intake_max = intake_max)
getMaxIntakeRate(params)["Cod", 1:5]
Set metabolic rate
Description
Sets the rate at which energy is used for metabolism and activity. You will
usually not need to call this function directly. Instead change the k,
ks and p species parameters with given_species_params(params) <- and
let mizer recalculate the metabolic rate for you. Call setMetabolicRate()
directly only if you want to impose a different functional form for the
size dependence of the metabolic rate. See
vignette("cheatsheet-changing-parameters") for a full explanation of when
to reach for which level of the model.
Usage
setMetabolicRate(object, metab = NULL, p = deprecated(), reset = FALSE, ...)
getMetabolicRate(params)
metab(params)
metab(params) <- value
Arguments
object |
A MizerParams object |
metab |
Optional. An array (species x size) holding the metabolic rate for each species at size. If not supplied, a default is set as described in the section "Setting metabolic rate". |
p |
|
reset |
If set to TRUE, then the metabolic rate will be reset to the value calculated from the species parameters, even if it was previously overwritten with a custom value. If set to FALSE (default) then a recalculation from the species parameters will take place only if no custom value has been set. |
... |
Unused |
params |
A MizerParams object |
value |
metab |
Value
setMetabolicRate(): A MizerParams object with updated metabolic rate.
getMetabolicRate() or equivalently metab(): A
ArraySpeciesBySize object (species x size) with the metabolic rate.
Setting metabolic rate
The metabolic rate is subtracted from the energy income rate to calculate
the rate at which energy is available for growth and reproduction, see
getEReproAndGrowth(). It is measured in grams/year.
If the metab argument is not supplied, then for each species the
metabolic rate k(w) for an individual of size w is set to
k(w) = k_s w^p + k w,
where k_s w^p represents the rate of standard metabolism and k w
is the rate at which energy is expended on activity and movement. The values
of k_s, p and k are taken from the ks, p and
k columns in the species parameter dataframe. If any of these
parameters are not supplied, the defaults are k = 0, p = n and
k_s = f_c h \alpha w_{mat}^{n-p},
where f_c is the critical feeding level taken from the fc column
in the species parameter data frame. If the critical feeding level is not
specified, a default of f_c = 0.2 is used.
If the metab slot has a comment and reset = FALSE, then a recalculation
from the species parameters is suppressed and a message is issued if the
recalculated values would differ from the stored ones.
See Also
Other functions for setting parameters:
gear_params(),
setExtDiffusion(),
setExtEncounter(),
setExtMort(),
setFishing(),
setInteraction(),
setMaxIntakeRate(),
setParams(),
setPredKernel(),
setReproduction(),
setSearchVolume(),
species_params(),
use_predation_diffusion()
Examples
# Inspect the current metabolic rate
getMetabolicRate(NS_params)["Cod", 1:5]
# Reset metabolic rate from species parameters
params <- setMetabolicRate(NS_params, reset = TRUE)
getMetabolicRate(params)["Cod", 1:5]
Set metadata for a model
Description
Setting metadata is particularly important for sharing your model with others. All metadata fields are optional and you can also add other fields of your own choosing. If you set a value for a field that already existed, the old value will be overwritten.
Usage
setMetadata(
params,
title = NULL,
description = NULL,
authors = NULL,
url = NULL,
doi = NULL,
...
)
getMetadata(params)
Arguments
params |
The MizerParams object for the model |
title |
A string with the title for the model |
description |
A string with a description of the model. This could for example contain information about any publications using the model. |
authors |
An author entry or a list of author entries, where each author
entry could either be just a name or could itself be a list with fields
like |
url |
A URL where more information about the model can be found. This could be a blog post on the mizer blog, for example. |
doi |
The digital object identifier for your model. To create a doi you can use online services like https://zenodo.org/ or https://figshare.com. |
... |
Additional metadata fields that you would like to add |
Details
In addition to the metadata fields you can set by hand, there are four fields that are set automatically by mizer:
-
mizer_versionThe version string of the mizer version under which the model was created or last upgraded. Can be compared to the current version which is obtained withpackageVersion("mizer"). The purpose of this field is that if the model is not working as expected in the current version of mizer, you can go back to the older version under which presumably it was working. -
extensionsA named vector of strings where each name is the name of and extension package needed to run the model and each value is a string giving the information that the remotes package needs to install the correct version of the extension package. This field is set by the extension packages. -
time_createdA POSIXct date-time object with the creation time. -
time_modifiedA POSIXct date-time object with the last modified time.
Setting the metadata with this function does not count as a modification of
the object, so the time_modified field will not be updated.
Value
setMetadata(): The MizerParams object with updated metadata
getMetadata(): A list with all metadata entries that have been set,
including at least
mizer_version, extensions, time_created and time_modified.
Examples
params <- setMetadata(NS_params,
title = "North Sea model",
description = "A multi-species model of the North Sea fish community.",
authors = list(list(name = "Finlay Scott", email = "finlay@example.com")),
my_own_filed = "something that doesn't fit elsewhere")
getMetadata(params)$title
getMetadata(params)$authors[[1]]$name
Set or change any model parameters
Description
This is a convenient wrapper function calling each of the following functions
See the Details section below for a discussion of how to use this function.
Usage
setParams(object, interaction = NULL, info_level = 3, ...)
Arguments
object |
A MizerParams object |
interaction |
Optional interaction matrix of the species (predator species x prey species). By default all entries are 1. See "Setting interaction matrix" section below. |
info_level |
Controls the amount of information messages that are shown. Higher levels lead to more messages. |
... |
Arguments passed on to
|
Details
If you are not happy with the assumptions that mizer makes by default about
the size-dependence of parameters, for example if you want to change one of
the allometric scaling assumptions, you can do this by providing your
choice as an array in the appropriate argument to setParams(). The
sections below discuss all the model functions that you can change this way.
Because of the way the R language works, setParams does not make the
changes to the params object that you pass to it but instead returns a new
params object. So to affect the change you call the function in the form
params <- setParams(params, ...).
Usually, if you are happy with the way mizer calculates the size-dependent parameters
from the species parameters and only want to change the values of some
species parameters, you would make those changes in the species_params data
frame contained in the params object using species_params<-().
Here is an example which assumes that
you have have a MizerParams object params in which you just want to change
the gamma parameter of the third species:
species_params(params)$gamma[[3]] <- 1000
Internally that will actually call setParams() to recalculate any of the
other parameters that are affected by the change in the species parameter.
setParams() will use the species parameters in the params object to
recalculate the values of all the parameter arrays except those for which you
have set custom values.
Value
A MizerParams object
Units in mizer
Mizer uses grams to measure weight, centimetres to measure lengths, and years to measure time.
Mizer is agnostic about whether abundances are given as
numbers per area,
numbers per volume or
total numbers for the entire study area.
You should make the choice most convenient for your application and then stick with it. If you make choice 1 or 2 you will also have to choose a unit for area or volume. Your choice will then determine the units for some of the parameters. This will be mentioned when the parameters are discussed in the sections below.
Your choice will also affect the units of the quantities you may want to
calculate with the model. For example, the yield will be in grams/year/m^2 in
case 1 if you choose m^2 as your measure of area, in grams/year/m^3 in case 2
if you choose m^3 as your unit of volume, or simply grams/year in case 3. The
same comment applies for other measures, like total biomass, which will be
grams/area in case 1, grams/volume in case 2 or simply grams in case 3. When
mizer puts units on axes in plots, it will choose the units appropriate for
case 3. So for example in plotBiomass() it gives the unit as grams.
You can convert between these choices. For example, if you use case 1, you
need to multiply with the area of the ecosystem to get the total quantity.
If you work with case 2, you need to multiply by both area and the thickness
of the productive layer. In that respect, case 2 is a bit cumbersome. The
function scaleModel() is useful to change the units you are using.
Setting interaction matrix
You do not need to specify an interaction matrix. If you do not, then the predator-prey interactions are purely determined by the size of predator and prey and totally independent of the species of predator and prey.
The interaction matrix \theta_{ij} modifies the interaction of each
pair of species in the model. This can be used for example to allow for
different spatial overlap among the species.
The values in the interaction matrix are used to scale the encountered food
and predation mortality (see on the website the section on predator-prey encounter rate
and on predation mortality).
The first index refers to the predator species and the second to the prey
species.
The interaction matrix is used when calculating the food encounter rate in
getEncounter() and the predation mortality rate in getPredMort(). Its
entries are dimensionless numbers. If all the values in the interaction
matrix are equal then predator-prey interactions are determined entirely by
size-preference.
This function checks that the supplied interaction matrix is valid and then
stores it in the interaction slot of the params object.
The order of the columns and rows of the interaction argument should be
the same as the order in the species params data frame in the params
object. If you supply a named array then the function will check the order
and message if it is different before ignoring the supplied dimnames. If
you supply only column names then these are also used as the row names. One
way of creating your own interaction
matrix is to enter the data using a spreadsheet program and saving it as a
.csv file. The data can then be read into R using the command read.csv().
The interaction of the species with the resource are set via a column
interaction_resource in the species_params data frame. By default this
column is set to all 1s.
Setting predation kernel
Kernel dependent on predator to prey size ratio
If the pred_kernel argument is not supplied, then this function sets a
predation kernel that depends only on the ratio of predator mass to prey
mass, not on the two masses independently. The shape of that kernel is then
determined by the pred_kernel_type column in species_params.
The default for pred_kernel_type is "lognormal". This will call the function
lognormal_pred_kernel() to calculate the predation kernel.
An alternative pred_kernel type is "box", implemented by the function
box_pred_kernel(), and "power_law", implemented by the function
power_law_pred_kernel(). These functions require certain species
parameters in the species_params data frame. For the lognormal kernel these
are beta and sigma, for the box kernel they are ppmr_min
and ppmr_max. They are explained in the help pages for the kernel
functions. Except for beta and sigma, no defaults are set for
these parameters. If they are missing from the species_params data frame then
mizer will issue an error message.
You can use any other string for pred_kernel_type. If for example you
choose "my" then you need to define a function my_pred_kernel that you can
model on the existing functions like lognormal_pred_kernel().
When using a kernel that depends on the predator/prey size ratio only, mizer
does not need to store the entire three dimensional array in the MizerParams
object. Such an array can be very big when there is a large number of size
bins. Instead, mizer only needs to store two two-dimensional arrays that hold
Fourier transforms of the feeding kernel function that allow the encounter
rate and the predation rate to be calculated very efficiently. However, if
you need the full three-dimensional array you can calculate it with the
getPredKernel() function.
Kernel dependent on both predator and prey size
If you want to work with a feeding kernel that depends on predator mass and prey mass independently, you can specify the full feeding kernel as a three-dimensional array (predator species x predator size x prey size).
You should use this option only if a kernel dependent only on the predator/prey mass ratio is not appropriate. Using a kernel dependent on predator/prey mass ratio only allows mizer to use fast Fourier transform methods to significantly reduce the running time of simulations.
The order of the predator species in pred_kernel should be the same
as the order in the species params dataframe in the params object. If you
supply a named array then the function will check the order and warn if it is
different.
Setting search volume
The search volume \gamma_i(w) of an individual of species i
and weight w multiplies the predation kernel when
calculating the encounter rate in getEncounter() and the
predation rate in getPredRate().
The name "search volume" is a bit misleading, because \gamma_i(w) does
not have units of volume. It is simply a parameter that determines the rate
of predation. Its units depend on your choice, see section "Units in mizer".
If you have chosen to work with total abundances, then it is a rate with units
1/year. If you have chosen to work with abundances per m^2 then it has units
of m^2/year. If you have chosen to work with abundances per m^3 then it has
units of m^3/year.
If the search_vol argument is not supplied, then the search volume is
set to
\gamma_i(w) = \gamma_i w^q_i.
The values of \gamma_i (the search volume at 1g) and q_i (the
allometric exponent of the search volume) are taken from the gamma and
q columns in the species parameter dataframe. If the gamma
column is not supplied in the species parameter dataframe, a default is
calculated by the get_gamma_default() function. If the q column is not
supplied, a default of lambda - 2 + n is used. Note that only
for predators of size w = 1 gram is the value of the species parameter
\gamma_i the same as the value of the search volume \gamma_i(w).
If the search_vol slot has a comment and reset = FALSE, then a
recalculation from the species parameters is suppressed and a message is
issued if the recalculated values would differ from the stored ones.
Setting maximum intake rate
The maximum intake rate h_i(w) of an individual of species i and
weight w determines the feeding level, calculated with
getFeedingLevel(). It is measured in grams/year.
If the intake_max argument is not supplied, then the maximum intake
rate is set to
h_i(w) = h_i w^{n_i}.
The values of h_i (the maximum intake rate of an individual of size 1
gram) and n_i (the allometric exponent for the intake rate) are taken
from the h and n columns in the species parameter dataframe. If
the h column is not supplied in the species parameter dataframe, it is
calculated by the get_h_default() function. If the n column is not
supplied, a default of n_i = 3/4 is used.
If h_i is set to Inf, fish of species i will consume all encountered
food.
If the intake_max slot has a comment and reset = FALSE, then a
recalculation from the species parameters is suppressed and a message is
issued if the recalculated values would differ from the stored ones.
Setting metabolic rate
The metabolic rate is subtracted from the energy income rate to calculate
the rate at which energy is available for growth and reproduction, see
getEReproAndGrowth(). It is measured in grams/year.
If the metab argument is not supplied, then for each species the
metabolic rate k(w) for an individual of size w is set to
k(w) = k_s w^p + k w,
where k_s w^p represents the rate of standard metabolism and k w
is the rate at which energy is expended on activity and movement. The values
of k_s, p and k are taken from the ks, p and
k columns in the species parameter dataframe. If any of these
parameters are not supplied, the defaults are k = 0, p = n and
k_s = f_c h \alpha w_{mat}^{n-p},
where f_c is the critical feeding level taken from the fc column
in the species parameter data frame. If the critical feeding level is not
specified, a default of f_c = 0.2 is used.
If the metab slot has a comment and reset = FALSE, then a recalculation
from the species parameters is suppressed and a message is issued if the
recalculated values would differ from the stored ones.
Setting external mortality rate
The external mortality is all the mortality that is not due to fishing or predation by predators included in the model. The external mortality could be due to predation by predators that are not explicitly included in the model (e.g. mammals or seabirds) or due to other causes like illness. It is a rate with units 1/year.
The ext_mort argument allows you to specify an external mortality rate
that depends on species and body size. You can see an example of this in
the Examples section of the help page for setExtMort().
If the ext_mort argument is not supplied, then the external mortality is
taken from the species parameters as
\mu_{ext.i}(w) = z_{0.i} + z_{ext.i} w^{d_i}.
The value of the constant z_0 for each species is taken from the z0
column of the species parameter data frame, if that column exists.
Otherwise it is calculated as
z_{0.i} = {\tt z0pre}_i\, w_{inf}^{\tt z0exp}.
Missing values of z_ext are set to 0 and missing values of d are set to
n - 1.
By default the power law is evaluated at the left bin edges w_j
(point sampling). If the bin_average entry of the second_order_w slot is
TRUE (see second_order_w()), then the z_{ext} w^d term is instead
replaced by its exact average over each bin [w_j, w_{j+1}],
\frac{z_{ext}}{\Delta w_j}\int_{w_j}^{w_{j+1}} w^d\, dw
= z_{ext}\,\frac{w_{j+1}^{d+1} - w_j^{d+1}}{(d+1)\,\Delta w_j},
(with the limiting form z_{ext}\ln(w_{j+1}/w_j)/\Delta w_j when
d = -1). This is the consistent choice in the finite-volume scheme,
where the external mortality multiplies the bin-averaged abundance. The
bin-averaging is applied only to the auto-calculated power-law default; a
user-supplied ext_mort array is left untouched.
Setting external encounter rate
The external encounter rate is the rate at which a predator encounters food that is not explicitly modelled. It is a rate with units mass/year.
The ext_encounter argument allows you to specify an external encounter rate
that depends on species and body size. You can see an example of this in
the Examples section of the help page for setExtEncounter().
If the ext_encounter argument is not supplied, then the external encounter
rate is calculated as a power law:
E_{ext.i}(w) = E_{ext.i}\, w^{n_i}.
The coefficient E_{ext.i} is taken from the E_ext column of the
species parameter data frame, which defaults to 0. The exponent n_i is
taken from the n column of the species parameter data frame.
If the ext_encounter slot has a comment and reset = FALSE, then a
recalculation from the species parameters is suppressed and a message is
issued if the recalculated values would differ from the stored ones.
Setting external diffusion rate
The external diffusion rate allows you to impose additional diffusion beyond the predation-driven diffusion that can be internally modelled by mizer.
The ext_diffusion argument allows you to specify a diffusion rate that
depends on species and body size.
If the ext_diffusion argument is not supplied, then the external diffusion
rate is calculated as a power law:
D_{ext.i}(w) = D_{ext.i}\, w^{n_i+1}.
The coefficient D_{ext.i} is taken from the D_ext column of the
species parameter data frame, which defaults to 0. The exponent
n_i + 1 uses the n column of the species parameter data frame.
If the ext_diffusion slot has a comment and reset = FALSE, then a
recalculation from the species parameters is suppressed and a message is
issued if the recalculated values would differ from the stored ones.
Setting reproduction
For each species and at each size, the proportion \psi of the
available energy
that is invested into reproduction is the product of two factors: the
proportion maturity of individuals that are mature and the proportion
repro_prop of the energy available to a mature individual that is
invested into reproduction. There is a size w_repro_max at which a typical
mature individual invests all of its available energy into reproduction.
This is not a hard ceiling on size: not all individuals are mature at
w_repro_max, and diffusion in the growth process allows some individuals to
grow beyond it, so fish larger than w_repro_max can exist. If you have not
specified the w_repro_max column in the species parameter data frame, then
the von Bertalanffy asymptotic size w_inf is used instead.
Maturity ogive
If the the proportion of individuals that are mature is not supplied via
the maturity argument, then it is set to a sigmoidal
maturity ogive that changes from 0 to 1 at around the maturity size:
{\tt maturity}(w) = \left[1+\left(\frac{w}{w_{mat}}\right)^{-U}\right]^{-1}.
(To avoid clutter, we are not showing the species index in the equations,
although each species has its own maturity ogive.)
The maturity weights are taken from the w_mat column of the
species_params data frame. Any missing maturity weights are set to 1/4 of the
asymptotic size in the w_inf column.
The exponent U determines the steepness of the maturity ogive. By
default it is chosen as U = 10, however this can be overridden by
including a column w_mat25 in the species parameter dataframe that
specifies the weight at which 25% of individuals are mature, which sets
U = \log(3) / \log(w_{mat} / w_{mat25}).
The sigmoidal function given above would strictly reach 0 only
asymptotically and thus have some (negligible) amount of reproduction at
arbitrarily small size.
For computational simplicity, any proportion smaller than
1e-8 is set to 0.
Investment into reproduction
If the the energy available to a mature individual that is
invested into reproduction is not supplied via the repro_prop argument,
it is set to the allometric form
{\tt repro\_prop}(w) =
\min\left(\left(\dfrac{w}{w_{\tt{repro\_max}}}\right)^{m-n},1\right).
Here n is the scaling exponent of the energy income rate. Hence
the exponent m determines the scaling of the investment into
reproduction for mature individuals. By default it is chosen to be
m = 1 so that the rate at which energy is invested into reproduction
scales linearly with the size. This default can be overridden by including a
column m in the species parameter dataframe. The sizes w_{repro\_max}
are taken from the w_repro_max column in the species parameter data frame,
if it exists, or otherwise from the w_inf column.
The total proportion of energy invested into reproduction of an individual
of size w is then
\psi(w) = {\tt maturity}(w){\tt repro\_prop}(w)
In mizer edition 1, at sizes above w_repro_max the value of \psi
is additionally forced to 1, so that all available energy is invested into
reproduction and growth stops. In edition 2 and above this forcing is not
applied, and \psi is determined entirely by the maturity ogive and the
reproductive proportion.
Reproductive efficiency
The reproductive efficiency \epsilon, i.e., the proportion of energy allocated to
reproduction that results in egg biomass, is set through the erepro
column in the species_params data frame. If that is not provided, the default
is set to 1 (which you will want to override). The offspring biomass divided
by the egg biomass gives the rate of egg production, returned by
getRDI():
R_{di} = \frac{\epsilon}{2 w_{min}} \int N(w) E_r(w) \psi(w) \, dw
Density dependence
The stock-recruitment relationship is an emergent phenomenon in mizer, with several sources of density dependence. Firstly, the amount of energy invested into reproduction depends on the energy income of the spawners, which is density-dependent due to competition for prey. Secondly, the proportion of larvae that grow up to recruitment size depends on the larval mortality, which depends on the density of predators, and on larval growth rate, which depends on density of prey.
Finally, to encode all the density dependence in the stock-recruitment
relationship that is not already included in the other two sources of density
dependence, mizer puts the the density-independent rate of egg production
through a density-dependence function. The result is returned by
getRDD(). The name of the density-dependence function is
specified by the RDD argument. The default is the Beverton-Holt
function BevertonHoltRDD(), which requires an R_max column
in the species_params data frame giving the maximum egg production rate. If
this column does not exist, it is initialised to Inf, leading to no
density-dependence. Other functions provided by mizer are
RickerRDD() and SheperdRDD() and you can easily use
these as models for writing your own functions.
Setting fishing
Gears
In mizer, fishing mortality is imposed on species by fishing gears. The
total per-capita fishing mortality (1/year) is obtained by summing over the
mortality from all gears,
\mu_{f.i}(w) = \sum_g F_{g,i}(w),
where the fishing mortality F_{g,i}(w) imposed by gear g on
species i at size w is calculated as:
F_{g,i}(w) = S_{g,i}(w) Q_{g,i} E_{g},
where S is the selectivity by species, gear and size, Q is the
catchability by species and gear and E is the fishing effort by gear.
Selectivity
The selectivity at size of each gear for each species is saved as a three
dimensional array (gear x species x size). Each entry has a range between 0
(that gear is not selecting that species at that size) to 1 (that gear is
selecting all individuals of that species of that size). This three
dimensional array can be specified explicitly via the selectivity
argument, but usually mizer calculates it from the gear_params slot of
the MizerParams object.
To allow the calculation of the selectivity array, the gear_params slot
must be a data frame with one row for each gear-species combination. So if
for example a gear can select three species, then that gear contributes three
rows to the gear_params data frame, one for each species it can select. The
data frame must have columns gear, holding the name of the gear, species,
holding the name of the species, and sel_func, holding the name of the
function that calculates the selectivity curve. Some selectivity functions
are included in the package: knife_edge(), sigmoid_length(),
double_sigmoid_length(), and sigmoid_weight().
Users are able to write their own size-based selectivity function. The first
argument to the function must be w and the function must return a vector of
the selectivity (between 0 and 1) at size.
Each selectivity function may have parameters. Values for these
parameters must be included as columns in the gear parameters data.frame.
The names of the columns must exactly match the names of the corresponding
arguments of the selectivity function. For example, the default selectivity
function is knife_edge() that a has sudden change of selectivity from 0 to 1
at a certain size. In its help page you can see that the knife_edge()
function has arguments w and knife_edge_size. The first argument, w, is
size (the function calculates selectivity at size). All selectivity functions
must have w as the first argument. The values for the other arguments must
be found in the gear parameters data.frame. So for the knife_edge()
function there should be a knife_edge_size column. Because knife_edge()
is the default selectivity function, the knife_edge_size argument has a
default value = w_mat.
The most commonly-used selectivity function is sigmoid_length(). It has a
smooth transition from 0 to 1 at a certain size. The sigmoid_length()
function has the two parameters l50 and l25 that are the lengths in cm at
which 50% or 25% of the fish are selected by the gear. If you choose this
selectivity function then the l50 and l25 columns must be included in the
gear parameters data.frame.
In case each species is only selected by one gear, the columns of the
gear_params data frame can alternatively be provided as columns of the
species_params data frame, if this is more convenient for the user to set
up. Mizer will then copy these columns over to create the gear_params data
frame when it creates the MizerParams object. However changing these columns
in the species parameter data frame later will not update the gear_params
data frame.
Catchability
Catchability is used as an additional factor to make the link between gear selectivity, fishing effort and fishing mortality. For example, it can be set so that an effort of 1 gives a desired fishing mortality. In this way effort can then be specified relative to a 'base effort', e.g. the effort in a particular year.
Catchability is stored as a two dimensional array (gear x species). This can
either be provided explicitly via the catchability argument, or the
information can be provided via a catchability column in the gear_params
data frame.
In the case where each species is selected by only a single gear, the
catchability column can also be provided in the species_params data
frame. Mizer will then copy this over to the gear_params data frame when
the MizerParams object is created.
Effort
The initial fishing effort is stored in the MizerParams object. If it is
not supplied, it is set to zero. The initial effort can be overruled when
the simulation is run with project(), where it is also possible to specify
an effort that varies through time.
See Also
Other functions for setting parameters:
gear_params(),
setExtDiffusion(),
setExtEncounter(),
setExtMort(),
setFishing(),
setInteraction(),
setMaxIntakeRate(),
setMetabolicRate(),
setPredKernel(),
setReproduction(),
setSearchVolume(),
species_params(),
use_predation_diffusion()
Set predation kernel
Description
You will usually not need to call this function directly. Instead change
the relevant species parameters (pred_kernel_type, and, depending on its
value, beta/sigma or ppmr_min/ppmr_max) with
given_species_params(params) <- and let mizer recalculate the predation
kernel for you. Call setPredKernel() directly only if you want to supply
the full kernel array yourself. See
vignette("cheatsheet-changing-parameters") for a full explanation of when
to reach for which level of the model.
Usage
setPredKernel(params, pred_kernel = NULL, reset = FALSE, ...)
getPredKernel(params)
pred_kernel(params)
pred_kernel(params) <- value
Arguments
params |
A MizerParams object |
pred_kernel |
Optional. An array (species x predator size x prey size) that holds the predation coefficient of each predator at size on each prey size. If not supplied, a default is set as described in section "Setting predation kernel". |
reset |
If set to TRUE, then the predation kernel will be reset to the value calculated from the species parameters, even if it was previously overwritten with a custom value. If set to FALSE (default) then a recalculation from the species parameters will take place only if no custom value has been set. |
... |
Unused |
value |
pred_kernel |
Details
The predation kernel determines the distribution of prey sizes that a
predator feeds on. It is used in getEncounter() when calculating
the rate at which food is encountered and in getPredRate() when
calculating the rate at which a prey is predated upon. The predation kernel
can be a function of the predator/prey size ratio or it can be a function of
the predator size and the prey size separately. Both types can be set up with
this function.
Value
setPredKernel(): A MizerParams object with updated predation kernel.
getPredKernel() or equivalently pred_kernel(): An array (predator
species x predator_size x prey_size)
Setting predation kernel
Kernel dependent on predator to prey size ratio
If the pred_kernel argument is not supplied, then this function sets a
predation kernel that depends only on the ratio of predator mass to prey
mass, not on the two masses independently. The shape of that kernel is then
determined by the pred_kernel_type column in species_params.
The default for pred_kernel_type is "lognormal". This will call the function
lognormal_pred_kernel() to calculate the predation kernel.
An alternative pred_kernel type is "box", implemented by the function
box_pred_kernel(), and "power_law", implemented by the function
power_law_pred_kernel(). These functions require certain species
parameters in the species_params data frame. For the lognormal kernel these
are beta and sigma, for the box kernel they are ppmr_min
and ppmr_max. They are explained in the help pages for the kernel
functions. Except for beta and sigma, no defaults are set for
these parameters. If they are missing from the species_params data frame then
mizer will issue an error message.
You can use any other string for pred_kernel_type. If for example you
choose "my" then you need to define a function my_pred_kernel that you can
model on the existing functions like lognormal_pred_kernel().
When using a kernel that depends on the predator/prey size ratio only, mizer
does not need to store the entire three dimensional array in the MizerParams
object. Such an array can be very big when there is a large number of size
bins. Instead, mizer only needs to store two two-dimensional arrays that hold
Fourier transforms of the feeding kernel function that allow the encounter
rate and the predation rate to be calculated very efficiently. However, if
you need the full three-dimensional array you can calculate it with the
getPredKernel() function.
Kernel dependent on both predator and prey size
If you want to work with a feeding kernel that depends on predator mass and prey mass independently, you can specify the full feeding kernel as a three-dimensional array (predator species x predator size x prey size).
You should use this option only if a kernel dependent only on the predator/prey mass ratio is not appropriate. Using a kernel dependent on predator/prey mass ratio only allows mizer to use fast Fourier transform methods to significantly reduce the running time of simulations.
The order of the predator species in pred_kernel should be the same
as the order in the species params dataframe in the params object. If you
supply a named array then the function will check the order and warn if it is
different.
Higher-order quadrature
When the predation kernel depends only on the predator/prey mass ratio, the
encounter and predation rates are evaluated as convolutions using the fast
Fourier transform. By default mizer point-samples the kernel at the grid
nodes, which is a first-order (rectangle-rule) quadrature. When the
bin_average entry of the second_order_w slot is TRUE, the kernel is
instead integrated over each logarithmic size bin when building the
Fourier-transformed kernels. This finite-volume consistent quadrature lifts
the encounter and predation rates towards second order at no extra runtime
cost, because the integration is performed once here and the rate functions
themselves are unchanged. The predation kernel is additionally averaged over
the prey bin (a trapezoid fold), so that the predation rate returned by
getPredRate() is the prey-bin average that the predation-mortality sink
needs to be second order. The default remains the first-order scheme so that
existing models are unaffected. Enable it with second_order_w(params) <- TRUE (see second_order_w()), which also turns on the other bin-averaged
rate quadratures so the whole model stays consistent. See the
vignette("fft") for the mathematical details.
See Also
Other functions for setting parameters:
gear_params(),
setExtDiffusion(),
setExtEncounter(),
setExtMort(),
setFishing(),
setInteraction(),
setMaxIntakeRate(),
setMetabolicRate(),
setParams(),
setReproduction(),
setSearchVolume(),
species_params(),
use_predation_diffusion()
Examples
## Set up a MizerParams object
params <- NS_params
## If you change predation kernel parameters after setting up a model,
# this will be used to recalculate the kernel
species_params(params)["Cod", "beta"] <- 200
## You can change to a different predation kernel type
species_params(params)$ppmr_max <- 4000
species_params(params)$ppmr_min <- 200
species_params(params)$pred_kernel_type <- "box"
plot(w_full(params), getPredKernel(params)["Cod", 100, ], type="l", log="x")
## If you need a kernel that depends also on prey size you need to define
# it yourself.
pred_kernel <- getPredKernel(params)
pred_kernel["Herring", , ] <- sweep(pred_kernel["Herring", , ], 2,
params@w_full, "*")
params<- setPredKernel(params, pred_kernel = pred_kernel)
Set own rate function to replace mizer rate function
Description
If the way mizer calculates a fundamental rate entering the model is
not flexible enough for you (for example if you need to introduce time
dependence) then you can write your own functions for calculating that
rate and use setRateFunction() to register it with mizer.
Usage
setRateFunction(params, rate, fun)
getRateFunction(params, rate)
other_params(params)
other_params(params) <- value
Arguments
params |
A MizerParams object |
rate |
Name of the rate for which a new function is to be set. |
fun |
Name of the function to use to calculate the rate. |
value |
A named list of user-defined parameters to store in
|
Details
At each time step during a simulation with the project() function, mizer
needs to calculate the instantaneous values of the various rates. By
default it calls the mizerRates() function which creates a list with the
following components:
-
encounterfrommizerEncounter() -
feeding_levelfrommizerFeedingLevel() -
pred_ratefrommizerPredRate() -
pred_mortfrommizerPredMort() -
f_mortfrommizerFMort() -
mortfrommizerMort() -
resource_mortfrommizerResourceMort() -
efrommizerEReproAndGrowth() -
e_reprofrommizerERepro() -
e_growthfrommizerEGrowth() -
diffusionfrommizerDiffusion() -
rdifrommizerRDI() -
rddfromBevertonHoltRDD()
For each of these you can substitute your own function. So for example if
you have written your own function for calculating the total mortality
rate and have called it myMort and have a mizer model stored in a
MizerParams object called params that you want to run with your new
mortality rate, then you would call
params <- setRateFunction(params, "Mort", "myMort")
In general if you want to replace a function mizerSomeRateFunc() with
a function myVersionOfThis() you would call
params <- setRateFunction(params, "SomeRateFunc", "myVersionOfThis")
In some extreme cases you may need to swap out the entire mizerRates()
function for your own function called myRates(). That you can do with
params <- setRateFunction(params, "Rates", "myRates")
Your new rate functions may need their own model parameters. These you
can store in other_params(params). For example
other_params(params)$my_param <- 42
Note that your own rate functions need to be defined in the global environment or in a package. If they are defined within a function then mizer will not find them.
Value
For setRateFunction(): An updated MizerParams object
For getRateFunction(): The name of the registered rate function for
the requested rate, or the list of all rate functions if called without
rate argument.
For other_params(): The user-defined parameters stored in
other_params(params), or NULL if none have been set. This excludes any
component-specific parameters stored via setComponent().
See Also
"Extending mizer":
vignette("extending-mizer", package = "mizer")
Other extension tools:
NOther(),
clearExtensionChain(),
coerceToExtensionClass(),
getRegisteredExtensions(),
initialNOther<-(),
recordExtension(),
registerExtension(),
registerExtensions(),
setComponent()
Set reproduction parameters
Description
Sets the proportion of the total energy available for reproduction and growth
that is invested into reproduction as a function of the size of the
individual and sets additional density dependence. You will usually not
need to call this function directly. Instead change the w_mat,
w_mat25, w_repro_max and m species parameters with
given_species_params(params) <- and let mizer recalculate the maturity
ogive and reproduction allocation for you. Call setReproduction()
directly only if you want to impose different functional forms for these.
See vignette("cheatsheet-changing-parameters") for a full explanation of
when to reach for which level of the model.
Usage
setReproduction(
params,
maturity = NULL,
repro_prop = NULL,
reset = FALSE,
RDD = NULL,
...
)
getMaturityProportion(params)
maturity(params)
maturity(params) <- value
getReproductionProportion(params)
repro_prop(params)
repro_prop(params) <- value
psi(params)
Arguments
params |
A MizerParams object |
maturity |
Optional. An array (species x size) that holds the proportion of individuals of each species at size that are mature. If not supplied, a default is set as described in the section "Setting reproduction". |
repro_prop |
Optional. An array (species x size) that holds the proportion of the energy available for growth and reproduction that a mature individual allocates to reproduction for each species at size. If not supplied, a default is set as described in the section "Setting reproduction". |
reset |
If set to TRUE, then both |
RDD |
The name of the function calculating the density-dependent
reproduction rate from the density-independent rate. Defaults to
" |
... |
Unused |
value |
The desired new value for the respective parameter. |
Value
setReproduction(): A MizerParams object with updated reproduction
parameters.
getMaturityProportion() or equivalently maturity():
An ArraySpeciesBySize object (species x size) that holds the proportion
of individuals of each species at size that are mature.
getReproductionProportion() or equivalently repro_prop():
An ArraySpeciesBySize object (species x size) that holds the proportion
of the energy available for growth and reproduction that a mature
individual allocates to reproduction for each species at size. For sizes
where the maturity proportion is zero, also the reproduction proportion is
returned as zero.
Setting reproduction
For each species and at each size, the proportion \psi of the
available energy
that is invested into reproduction is the product of two factors: the
proportion maturity of individuals that are mature and the proportion
repro_prop of the energy available to a mature individual that is
invested into reproduction. There is a size w_repro_max at which a typical
mature individual invests all of its available energy into reproduction.
This is not a hard ceiling on size: not all individuals are mature at
w_repro_max, and diffusion in the growth process allows some individuals to
grow beyond it, so fish larger than w_repro_max can exist. If you have not
specified the w_repro_max column in the species parameter data frame, then
the von Bertalanffy asymptotic size w_inf is used instead.
Maturity ogive
If the the proportion of individuals that are mature is not supplied via
the maturity argument, then it is set to a sigmoidal
maturity ogive that changes from 0 to 1 at around the maturity size:
{\tt maturity}(w) = \left[1+\left(\frac{w}{w_{mat}}\right)^{-U}\right]^{-1}.
(To avoid clutter, we are not showing the species index in the equations,
although each species has its own maturity ogive.)
The maturity weights are taken from the w_mat column of the
species_params data frame. Any missing maturity weights are set to 1/4 of the
asymptotic size in the w_inf column.
The exponent U determines the steepness of the maturity ogive. By
default it is chosen as U = 10, however this can be overridden by
including a column w_mat25 in the species parameter dataframe that
specifies the weight at which 25% of individuals are mature, which sets
U = \log(3) / \log(w_{mat} / w_{mat25}).
The sigmoidal function given above would strictly reach 0 only
asymptotically and thus have some (negligible) amount of reproduction at
arbitrarily small size.
For computational simplicity, any proportion smaller than
1e-8 is set to 0.
Investment into reproduction
If the the energy available to a mature individual that is
invested into reproduction is not supplied via the repro_prop argument,
it is set to the allometric form
{\tt repro\_prop}(w) =
\min\left(\left(\dfrac{w}{w_{\tt{repro\_max}}}\right)^{m-n},1\right).
Here n is the scaling exponent of the energy income rate. Hence
the exponent m determines the scaling of the investment into
reproduction for mature individuals. By default it is chosen to be
m = 1 so that the rate at which energy is invested into reproduction
scales linearly with the size. This default can be overridden by including a
column m in the species parameter dataframe. The sizes w_{repro\_max}
are taken from the w_repro_max column in the species parameter data frame,
if it exists, or otherwise from the w_inf column.
The total proportion of energy invested into reproduction of an individual
of size w is then
\psi(w) = {\tt maturity}(w){\tt repro\_prop}(w)
In mizer edition 1, at sizes above w_repro_max the value of \psi
is additionally forced to 1, so that all available energy is invested into
reproduction and growth stops. In edition 2 and above this forcing is not
applied, and \psi is determined entirely by the maturity ogive and the
reproductive proportion.
Reproductive efficiency
The reproductive efficiency \epsilon, i.e., the proportion of energy allocated to
reproduction that results in egg biomass, is set through the erepro
column in the species_params data frame. If that is not provided, the default
is set to 1 (which you will want to override). The offspring biomass divided
by the egg biomass gives the rate of egg production, returned by
getRDI():
R_{di} = \frac{\epsilon}{2 w_{min}} \int N(w) E_r(w) \psi(w) \, dw
Density dependence
The stock-recruitment relationship is an emergent phenomenon in mizer, with several sources of density dependence. Firstly, the amount of energy invested into reproduction depends on the energy income of the spawners, which is density-dependent due to competition for prey. Secondly, the proportion of larvae that grow up to recruitment size depends on the larval mortality, which depends on the density of predators, and on larval growth rate, which depends on density of prey.
Finally, to encode all the density dependence in the stock-recruitment
relationship that is not already included in the other two sources of density
dependence, mizer puts the the density-independent rate of egg production
through a density-dependence function. The result is returned by
getRDD(). The name of the density-dependence function is
specified by the RDD argument. The default is the Beverton-Holt
function BevertonHoltRDD(), which requires an R_max column
in the species_params data frame giving the maximum egg production rate. If
this column does not exist, it is initialised to Inf, leading to no
density-dependence. Other functions provided by mizer are
RickerRDD() and SheperdRDD() and you can easily use
these as models for writing your own functions.
See Also
Other functions for setting parameters:
gear_params(),
setExtDiffusion(),
setExtEncounter(),
setExtMort(),
setFishing(),
setInteraction(),
setMaxIntakeRate(),
setMetabolicRate(),
setParams(),
setPredKernel(),
setSearchVolume(),
species_params(),
use_predation_diffusion()
Examples
# Plot maturity and reproduction ogives for Cod in North Sea model
maturity <- getMaturityProportion(NS_params)["Cod", ]
repro_prop <- getReproductionProportion(NS_params)["Cod", ]
df <- data.frame(Size = w(NS_params),
Reproduction = repro_prop,
Maturity = maturity,
Total = maturity * repro_prop)
dff <- reshape2::melt(df, id.vars = "Size",
variable.name = "Type",
value.name = "Proportion")
library(ggplot2)
ggplot(dff) + geom_line(aes(x = Size, y = Proportion, colour = Type))
Set resource dynamics
Description
Sets the intrinsic resource birth rate and the intrinsic resource carrying
capacity as well as the name of the function used to simulate the resource
dynamics. By default, the birth rate and the carrying capacity are changed
together in such a way that the resource replenishes at the same rate at
which it is consumed. So you should only provide either the
resource_rate or the resource_capacity (or resource_level) because
the other is determined by the requirement that the resource replenishes
at the same rate at which it is consumed.
Usage
setResource(
params,
resource_rate = NULL,
resource_capacity = NULL,
resource_level = NULL,
resource_dynamics = NULL,
lambda = resource_params(params)[["lambda"]],
n = resource_params(params)[["n"]],
w_pp_cutoff = resource_params(params)[["w_pp_cutoff"]],
balance = NULL,
reset = FALSE,
...
)
resource_rate(params)
resource_rate(params, balance = NULL) <- value
resource_capacity(params)
resource_capacity(params, balance = NULL) <- value
resource_level(params)
resource_level(params, balance = NULL) <- value
resource_dynamics(params)
resource_dynamics(params, balance = NULL) <- value
Arguments
params |
A MizerParams object |
resource_rate |
Optional. A vector of per-capita resource birth rate for each size class or a single number giving the coefficient in the power-law for this rate, see "Setting resource dynamics" below. Must be strictly positive. |
resource_capacity |
Optional. Vector of resource intrinsic carrying capacities or coefficient in the power-law for the capacity, see "Setting resource dynamics" below. The resource capacity must not be smaller than the resource abundance. |
resource_level |
Optional. The ratio between the current resource number
density and the resource capacity. Either a number used at all sizes or a
vector specifying a value for each size. Must be greater than 0 and at
most 1,
except at sizes where the resource is zero, where it can be |
resource_dynamics |
Optional. Name of the function that determines the resource dynamics by calculating the resource spectrum at the next time step from the current state. |
lambda |
Used to set power-law exponent for resource capacity if the
|
n |
Used to set power-law exponent for resource rate if the
|
w_pp_cutoff |
The upper cut off size of the resource spectrum power law
used when |
balance |
By default, if possible, the resource parameters are set so that the resource replenishes at the same rate at which it is consumed. In this case you should only specify either the resource rate or the resource capacity (or resource level) because the other is then determined automatically. Set to FALSE if you do not want the balancing. |
reset |
If set to TRUE, then the resource capacity and birth rate will be reset to the values calculated from the resource parameters, even if they were previously overwritten with custom values. If set to FALSE (default) then a recalculation from the resource parameters will take place only if no custom values have been set. |
... |
Unused |
value |
The desired new value for the respective parameter. |
Details
You would usually set the resource dynamics only after having finished the
calibration of the steady state. Then setting the resource dynamics with
this function will preserve that steady state, unless you explicitly
choose to set balance = FALSE. Your choice of the resource dynamics only
affects the dynamics around the steady state. The higher the resource rate
or the lower the resource capacity the less sensitive the model will be to
changes in the competition for resource.
If you provide the resource_level then that sets the resource_capacity
to the current resource number density divided by the resource level. So
in that case you should not specify resource_capacity as well.
If you provide none of the arguments resource_level, resource_rate or
resource_capacity, and you do not change any of the resource parameters,
then the resource rate is kept at its previous value and, when balancing, the
capacity is recalculated from it. If instead you change one of the resource
parameters (kappa, lambda, n or w_pp_cutoff) or set reset = TRUE,
the rate and capacity are recalculated from the resource parameters (and then
balanced, unless balance = FALSE).
Value
setResource: A MizerParams object with updated resource parameters
A vector with the intrinsic resource birth rate for each size class.
A vector with the intrinsic resource capacity for each size class.
A vector with the ratio between the current resource number density and the resource capacity for each size class.
The name of the function that determines the resource dynamics.
Setting resource dynamics
The resource_dynamics argument allows you to choose the resource dynamics
function. By default, mizer uses a semichemostat model to describe the
resource dynamics in each size class independently. This semichemostat
dynamics is implemented by the function resource_semichemostat(). You can
change that to use a logistic model implemented by resource_logistic() or
you can use resource_constant() which keeps the resource constant or you
can write your own function.
Both the resource_semichemostat() and the resource_logistic() dynamics
are parametrised in terms of a size-dependent birth rate r_R(w) and a
size-dependent capacity c_R. The help pages of these functions give
the details.
The resource_rate argument can be a vector (with the same length as
w_full(params)) specifying the intrinsic resource birth rate for each size
class. Alternatively it can be a single number that is used as the
coefficient in a power law: then the intrinsic birth rate r_R(w) at
size w is set to
r_R(w) = r_R w^{n-1}.
The power-law exponent n is taken from the n argument.
The resource_capacity argument can be a vector specifying the intrinsic
resource carrying capacity for each size class. Alternatively it can be a
single number that is used as the coefficient in a truncated power
law: then the intrinsic carrying capacity c_R(w) at size w
is set to
c_R(w) = c_R\, w^{-\lambda}
for all w less than w_pp_cutoff and zero for larger sizes.
The power-law exponent \lambda is taken from the lambda argument.
The values for lambda, n and w_pp_cutoff are stored in a list
in the resource_params slot of the MizerParams object so that they can be
re-used automatically in the future. If you specify resource_rate or
resource_capacity as a single number, that coefficient is likewise stored,
as r_pp and kappa respectively. That list can be accessed with
resource_params().
See Also
Examples
params <- NS_params
resource_dynamics(params)
resource_dynamics(params) <- "resource_constant"
Alias for setBevertonHolt()
Description
An alias provided for backward compatibility with mizer version <= 2.0.4
Usage
setRmax(params, erepro, R_max, reproduction_level, ...)
Arguments
params |
A MizerParams object |
erepro |
Reproductive efficiency for each species. See details. |
R_max |
Maximum reproduction rate. See details. |
reproduction_level |
Sets |
... |
Unused
|
Details
With Beverton-Holt density dependence the relation between the energy
invested into reproduction and the number of eggs hatched is determined
by two parameters: the reproductive efficiency erepro and the maximum
reproduction rate R_max.
If no maximum is imposed on the reproduction rate
(R_{max} = \infty) then the resulting density-independent
reproduction rate R_{di} is proportional
to the total rate E_R at which energy is invested into reproduction,
R_{di} = \frac{\rm{erepro}}{2 w_{min}} E_R,
where the proportionality factor is given by the reproductive efficiency
erepro divided by the egg size w_min to convert energy to egg number and
divided by 2 to account for the two sexes.
Imposing a finite maximum reproduction rate R_{max} leads to a
non-linear relationship between energy invested and eggs hatched. This
density-dependent reproduction rate R_{dd} is given as
R_{dd} = R_{di}
\frac{R_{max}}{R_{di} + R_{max}}.
(All quantities in the above equations are species-specific but we dropped the species index for simplicity.)
The following plot illustrates the Beverton-Holt density dependence in the
reproduction rate for two different choices of parameters.
This plot shows that a given energy E_R invested into reproduction can
lead to the same reproduction rate R_{dd} with different choices
of the parameters R_max and erepro. R_max determines the asymptote of
the curve and erepro its initial slope. A higher R_max coupled with a
lower erepro (black curves) can give the same value as a lower R_max
coupled with a higher erepro (blue curves).
For the given initial state in the MizerParams object params one can
calculate the energy E_R that is invested into reproduction by the
mature individuals and the reproduction rate R_{dd} that is
required to keep the egg abundance constant. These two values determine the
location of the black dot in the above graph. You then only need one
parameter to select one curve from the family of Beverton-Holt curves going
through that point. This parameter can be erepro or R_max. Instead of
R_max you can alternatively specify the reproduction_level which is the
ratio between the density-dependent reproduction rate R_{dd} and
the maximal reproduction rate R_{max}.
If you do not provide a value for any of the reproduction parameter
arguments, then erepro will be set to the value it has in the current
species parameter data frame. If you do provide one of the reproduction
parameters, this can be either a vector with one value for each
species, or a named vector where the names determine which species are
affected, or a single unnamed value that is then used for all species. Any
species for which the given value is NA will remain unaffected.
The values for R_max must be larger than R_{dd} and can range
up to Inf. If a smaller value is requested a warning is issued and the
value is increased to the value required for a reproduction level of 0.99.
The values for the reproduction_level must be non-negative and
less than 1. The values for erepro must be large enough to allow the
required reproduction rate. If a smaller value is requested a warning is
issued and the value is increased to the smallest possible value. The values
for erepro should also be smaller than 1 to be physiologically sensible,
but this is not enforced by the function.
As can be seen in the graph above, choosing a lower value for R_max or a
higher value for erepro means that near the steady state the reproduction
will be less sensitive to a change in the energy invested into reproduction
and hence less sensitive to changes in the spawning stock biomass or its
energy income. As a result the species will also be less sensitive to
fishing, leading to a higher F_MSY.
Value
A MizerParams object
Examples
params <- NS_params
species_params(params)$erepro
# Attempting to set the same erepro for all species
params <- setBevertonHolt(params, erepro = 0.1)
t(species_params(params)[, c("erepro", "R_max")])
# Setting erepro for some species
params <- setBevertonHolt(params, erepro = c("Gurnard" = 0.6, "Plaice" = 0.95))
t(species_params(params)[, c("erepro", "R_max")])
# Setting R_max
R_max <- 1e17 * species_params(params)$w_max^-1
params <- setBevertonHolt(NS_params, R_max = R_max)
t(species_params(params)[, c("erepro", "R_max")])
# Setting reproduction_level
params <- setBevertonHolt(params, reproduction_level = 0.3)
t(species_params(params)[, c("erepro", "R_max")])
Set search volume
Description
You will usually not need to call this function directly. Instead change
the gamma and q species parameters with given_species_params(params) <-
and let mizer recalculate the search volume for you. Call setSearchVolume()
directly only if you want to impose a different functional form for the size
dependence of the search volume. See vignette("cheatsheet-changing-parameters")
for a full explanation of when to reach for which level of the model.
Usage
setSearchVolume(params, search_vol = NULL, reset = FALSE, ...)
getSearchVolume(params)
search_vol(params)
search_vol(params) <- value
Arguments
params |
MizerParams |
search_vol |
Optional. An array (species x size) holding the search volume for each species at size. If not supplied, a default is set as described in the section "Setting search volume". |
reset |
If set to TRUE, then the search volume will be reset to the value calculated from the species parameters, even if it was previously overwritten with a custom value. If set to FALSE (default) then a recalculation from the species parameters will take place only if no custom value has been set. |
... |
Unused |
value |
search_vol |
Value
setSearchVolume(): A MizerParams object with updated search volume.
getSearchVolume() or equivalently search_vol(): A
ArraySpeciesBySize object (species x size) holding the search volume.
Setting search volume
The search volume \gamma_i(w) of an individual of species i
and weight w multiplies the predation kernel when
calculating the encounter rate in getEncounter() and the
predation rate in getPredRate().
The name "search volume" is a bit misleading, because \gamma_i(w) does
not have units of volume. It is simply a parameter that determines the rate
of predation. Its units depend on your choice, see section "Units in mizer".
If you have chosen to work with total abundances, then it is a rate with units
1/year. If you have chosen to work with abundances per m^2 then it has units
of m^2/year. If you have chosen to work with abundances per m^3 then it has
units of m^3/year.
If the search_vol argument is not supplied, then the search volume is
set to
\gamma_i(w) = \gamma_i w^q_i.
The values of \gamma_i (the search volume at 1g) and q_i (the
allometric exponent of the search volume) are taken from the gamma and
q columns in the species parameter dataframe. If the gamma
column is not supplied in the species parameter dataframe, a default is
calculated by the get_gamma_default() function. If the q column is not
supplied, a default of lambda - 2 + n is used. Note that only
for predators of size w = 1 gram is the value of the species parameter
\gamma_i the same as the value of the search volume \gamma_i(w).
If the search_vol slot has a comment and reset = FALSE, then a
recalculation from the species parameters is suppressed and a message is
issued if the recalculated values would differ from the stored ones.
See Also
Other functions for setting parameters:
gear_params(),
setExtDiffusion(),
setExtEncounter(),
setExtMort(),
setFishing(),
setInteraction(),
setMaxIntakeRate(),
setMetabolicRate(),
setParams(),
setPredKernel(),
setReproduction(),
species_params(),
use_predation_diffusion()
Examples
# Inspect the current search volume
getSearchVolume(NS_params)["Cod", 1:5]
# Double the search volume for all species
sv <- getSearchVolume(NS_params) * 2
params <- setSearchVolume(NS_params, search_vol = sv)
getSearchVolume(params)["Cod", 1:5]
Deprecated function for setting up parameters for a community-type model
Description
This function has been deprecated in favour of the function
newCommunityParams() that sets better default values.
Usage
set_community_model(
max_w = 1e+06,
min_w = 0.001,
min_w_pp = 1e-10,
z0 = 0.1,
alpha = 0.2,
h = 10,
beta = 100,
sigma = 2,
q = 0.8,
n = 2/3,
kappa = 1000,
lambda = 2 + q - n,
f0 = 0.7,
r_pp = 10,
gamma = NA,
knife_edge_size = 1000,
knife_is_min = TRUE,
recruitment = kappa * min_w^-lambda,
rec_mult = 1,
...
)
Arguments
max_w |
The maximum size of the community. The |
min_w |
The minimum size of the community. Default value is 1e-3. |
min_w_pp |
The smallest size of the resource spectrum. |
z0 |
The background mortality of the community. Default value is 0.1. |
alpha |
The assimilation efficiency of the community. Default value 0.2 |
h |
The maximum food intake rate. Default value is 10. |
beta |
The preferred predator prey mass ratio. Default value is 100. |
sigma |
The width of the prey preference. Default value is 2.0. |
q |
The search volume exponent. Default value is 0.8. |
n |
The scaling of the intake. Default value is 2/3. |
kappa |
The carrying capacity of the resource spectrum. Default value is 1000. |
lambda |
The exponent of the resource spectrum. Default value is 2 + q - n. |
f0 |
The average feeding level of individuals who feed on a power-law
spectrum. This value is used to calculate the search rate parameter
|
r_pp |
Growth rate parameter for the resource spectrum. Default value is 10. |
gamma |
Volumetric search rate. Estimated using |
knife_edge_size |
The size at the edge of the knife-selectivity function. Default value is 1000. |
knife_is_min |
Is the knife-edge selectivity function selecting above (TRUE) or below (FALSE) the edge. Default is TRUE. |
recruitment |
The constant recruitment in the smallest size class of the
community spectrum. This should be set so that the community spectrum
continues the resource spectrum. Default value = |
rec_mult |
Additional multiplier for the constant recruitment. Default value is 1. |
... |
Other arguments to pass to the |
Details
This functions creates a MizerParams object so that
community-type models can be easily set up and run. A community model has
several features that distinguish it from the food-web type models. Only one
'species' is resolved, i.e. one 'species' is used to represent the whole
community. The resource spectrum only extends to the start of the community
spectrum. Recruitment to the smallest size in the community spectrum is
constant and set by the user. As recruitment is constant, the proportion of
energy invested in reproduction (the slot psi of the returned
MizerParams object) is set to 0. Standard metabolism has been turned
off (the parameter ks is set to 0). Consequently, the growth rate is
now determined solely by the assimilated food (see the package vignette for
more details).
The function has many arguments, all of which have default values. The main
arguments that the users should be concerned with are z0,
recruitment, alpha and f0 as these determine the average
growth rate of the community.
Fishing selectivity is modelled as a knife-edge function with one parameter,
knife_edge_size, which determines the size at which species are
selected.
The resulting MizerParams object can be projected forward using
project() like any other MizerParams object. When projecting
the community model it may be necessary to keep a small time step size
dt of around 0.1 to avoid any instabilities with the solver. You can
check for these numerical instabilities by plotting the biomass or abundance
through time after the projection.
Value
An object of type MizerParams
References
K. H. Andersen,J. E. Beyer and P. Lundberg, 2009, Trophic and individual efficiencies of size-structured communities, Proceedings of the Royal Society, 276, 109-114
Examples
params <- set_community_model(f0=0.7, z0=0.2, recruitment=3e7)
# This is now achieved with
params <- newCommunityParams(f0 = 0.7, z0 = 0.2)
sim <- project(params, effort = 0, t_max = 100, dt=0.1)
plotBiomass(sim)
plotSpectra(sim)
Deprecated obsolete function for setting up multispecies parameters
Description
This function has been deprecated in favour of the function
newMultispeciesParams() that sets better default values.
This wrapper keeps the legacy defaults and also fills in several columns in
species_params if they are missing, using the same rules as older mizer
versions.
Usage
set_multispecies_model(
species_params,
interaction = matrix(1, nrow = nrow(species_params), ncol = nrow(species_params)),
min_w_pp = 1e-10,
min_w = 0.001,
max_w = NULL,
no_w = 100,
n = 2/3,
q = 0.8,
f0 = 0.6,
kappa = 1e+11,
lambda = 2 + q - n,
r_pp = 10,
...
)
Arguments
species_params |
A data frame of species-specific parameter values. |
interaction |
Optional interaction matrix of the species (predator species x prey species). By default all entries are 1. See "Setting interaction matrix" section below. |
min_w_pp |
The smallest size of the resource spectrum. By default this is set to the smallest value at which any of the consumers can feed. |
min_w |
Sets the size of the eggs of all species for which this is not
given in the |
max_w |
The largest size of the consumer spectrum. By default this is
set to the largest |
no_w |
The number of size bins in the consumer spectrum. |
n |
The allometric growth exponent. This can be overruled for individual
species by including a |
q |
Allometric exponent of search volume |
f0 |
Expected average feeding level. Used to set |
kappa |
The coefficient |
lambda |
Used to set power-law exponent for resource capacity if the
|
r_pp |
|
... |
Further arguments passed to |
Details
If species_params contains a w_inf column then it is copied to w_max.
If max_w is not supplied then it is set to 1.1 * max(species_params$w_max).
The supplied min_w_pp is shifted up by one grid step before being passed
to newMultispeciesParams() to compensate for the fact that newer mizer
versions extend the full size grid below min_w_pp.
Missing legacy columns in species_params are filled as follows:
gear = species, k = 0, alpha = 0.6, erepro = 1,
sel_func = "knife_edge", knife_edge_size = w_mat if needed,
catchability = 1, ks = h * 0.2, and m = 1.
If h is missing it is calculated from k_vb, alpha, f0 and w_max.
If gamma is missing it is calculated from f0, h, beta, sigma,
lambda and kappa.
Value
A MizerParams object
Set a species parameter to a default value
Description
If the species parameter does not yet exist in the species parameter data
frame, then create it and fill it with the default. Otherwise use the default
only to fill in any NAs. Optionally gives a message if the parameter
did not already exist. The signal has class info_about_default.
Usage
set_species_param_default(object, parname, default, message = NULL)
Arguments
object |
Either a MizerParams object or a species parameter data frame |
parname |
A string with the name of the species parameter to set |
default |
A single default value or a vector with one default value for each species |
message |
A string with a message to be issued when the parameter did not already exist |
Value
The object with an updated column in the species params data frame.
Deprecated function for setting up parameters for a trait-based model
Description
This function has been deprecated in favour of the function
newTraitParams() that sets better default values.
Usage
set_trait_model(
no_sp = 10,
min_w_inf = 10,
max_w_inf = 1e+05,
no_w = 100,
min_w = 0.001,
max_w = max_w_inf * 1.1,
min_w_pp = 1e-10,
w_pp_cutoff = 1,
k0 = 50,
n = 2/3,
p = 0.75,
q = 0.9,
eta = 0.25,
r_pp = 4,
kappa = 0.005,
lambda = 2 + q - n,
alpha = 0.6,
ks = 4,
z0pre = 0.6,
h = 30,
beta = 100,
sigma = 1.3,
f0 = 0.5,
gamma = NA,
knife_edge_size = 1000,
gear_names = "knife_edge_gear",
...
)
Arguments
no_sp |
The number of species in the model. The default value is 10. The more species, the longer takes to run. |
min_w_inf |
The asymptotic size of the smallest species in the community. |
max_w_inf |
The asymptotic size of the largest species in the community. |
no_w |
The number of size bins in the community spectrum. |
min_w |
The smallest size of the community spectrum. |
max_w |
Maximum size of the consumer size grid passed to
|
min_w_pp |
Smallest size on the resource size grid passed to
|
w_pp_cutoff |
The cut off size of the resource spectrum. Default value is 1. |
k0 |
Multiplier for the maximum recruitment. Default value is 50. |
n |
Scaling of the intake. Default value is 2/3. |
p |
Scaling of the standard metabolism. Default value is 0.75. |
q |
Exponent of the search volume. Default value is 0.9. |
eta |
Factor to calculate |
r_pp |
Growth rate parameter for the resource spectrum. Default value is 4. |
kappa |
Coefficient in abundance power law. Default value is 0.005. |
lambda |
Exponent of the abundance power law. Default value is (2+q-n). |
alpha |
The assimilation efficiency of the community. The default value is 0.6 |
ks |
Standard metabolism coefficient. Default value is 4. |
z0pre |
The coefficient of the background mortality of the community. z0 = z0pre * w_inf ^ (n-1). The default value is 0.6. |
h |
Maximum food intake rate. Default value is 30. |
beta |
Preferred predator prey mass ratio. Default value is 100. |
sigma |
Width of prey size preference. Default value is 1.3. |
f0 |
Expected average feeding level. Used to set |
gamma |
Volumetric search rate. Estimated using |
knife_edge_size |
The minimum size at which the gear or gears select species. Must be of length 1 or no_sp. |
gear_names |
The names of the fishing gears. A character vector, the same length as the number of species. Default is 1 - no_sp. |
... |
Other arguments to pass to the |
Details
This functions creates a MizerParams object so that trait-based-type
models can be easily set up and run. The trait-based size spectrum model can
be derived as a simplification of the general size-based model used in
mizer. The species-specific parameters are the same for all species,
except for
the asymptotic size, which is considered the most important trait
characterizing a species. Other parameters are related to the asymptotic
size. For example, the size at maturity is given by w_max * eta,
where eta is
the same for all species. For the trait-based model the number of species is
not important. For applications of the trait-based model see Andersen &
Pedersen (2010). See the mizer vignette for more details and examples
of the trait-based model.
The function has many arguments, all of which have default values. Of particular interest to the user are the number of species in the model and the minimum and maximum asymptotic sizes. The asymptotic sizes of the species are spread evenly on a logarithmic scale within this range.
The stock recruitment relationship is the default Beverton-Holt style. The
maximum recruitment is calculated using equilibrium theory (see Andersen &
Pedersen, 2010) and a multiplier, k0. Users should adjust k0 to
get the spectra they want.
The factor for the search volume, gamma, is calculated using the
expected feeding level, f0.
Fishing selectivity is modelled as a knife-edge function with one parameter,
knife_edge_size, which is the size at which species are selected. Each
species can either be fished by the same gear (knife_edge_size has a
length of 1) or by a different gear (the length of knife_edge_size has
the same length as the number of species and the order of selectivity size is
that of the asymptotic size).
The resulting MizerParams object can be projected forward using
project like any other MizerParams object. When projecting
the community model it may be necessary to reduce dt to 0.1 to avoid
any instabilities with the solver. You can check this by plotting the biomass
or abundance through time after the projection.
Value
An object of type MizerParams
References
K. H. Andersen and M. Pedersen, 2010, Damped trophic cascades driven by fishing in model marine ecosystems. Proceedings of the Royal Society V, Biological Sciences, 1682, 795-802.
Length based sigmoid selectivity function
Description
A sigmoid shaped selectivity function. Based on two parameters l25 and
l50 which determine the length at which 25% and 50% of the stock is
selected respectively.
Usage
sigmoid_length(w, l25, l50, species_params, ...)
Arguments
w |
Vector of sizes. |
l25 |
the length which gives a selectivity of 25%. |
l50 |
the length which gives a selectivity of 50%. |
species_params |
A list with the species params for the current species.
Used to get at the length-weight parameters |
... |
Unused |
Details
You would not usually call this function directly. Instead, set the sel_func
column in gear_params() to "sigmoid_length" and provide the l25 and
l50 values as additional columns. setFishing() will then call this
function automatically when calculating the selectivity array.
The selectivity is given by the logistic function
S(l) = \frac{1}{1 + \exp\left(\log(3)\frac{l50 -l}{l50 - l25}\right)}
As the mizer model is weight based, and this
selectivity function is length based, it uses the
length-weight parameters a and b to convert between length and weight
l = \left(\frac{w}{a}\right)^{1/b}
Value
Vector of selectivities at the given sizes.
See Also
gear_params() for setting the selectivity parameters.
Other selectivity functions:
double_sigmoid_length(),
knife_edge(),
sigmoid_weight()
Examples
# Selectivity at weight given l25 = 10 cm, l50 = 15 cm
# using length-weight parameters a = 0.01, b = 3
sp <- list(a = 0.01, b = 3)
w <- c(1, 10, 100, 500, 1000)
sigmoid_length(w, l25 = 10, l50 = 15, species_params = sp)
Weight based sigmoidal selectivity function
Description
A sigmoidal selectivity function with 50% selectivity at
weight sigmoidal_weight =w_{\text{sigmoid}} and width sigmoidal_sigma =\sigma.
S(w) = \left(1 + \left(\frac{w}{w_{\text{sigmoid}}}\right)^{-\sigma}\right)^{-1}
Usage
sigmoid_weight(w, sigmoidal_weight, sigmoidal_sigma, ...)
Arguments
w |
Vector of sizes. |
sigmoidal_weight |
The weight at which selectivity is 50%. |
sigmoidal_sigma |
The width of the selection function. |
... |
Unused |
Details
You would not usually call this function directly. Instead, set the sel_func
column in gear_params() to "sigmoid_weight" and provide sigmoidal_weight
and sigmoidal_sigma as additional columns. setFishing() will then call
this function automatically when calculating the selectivity array.
Value
Vector of selectivities at the given sizes.
See Also
gear_params() for setting the selectivity parameters.
Other selectivity functions:
double_sigmoid_length(),
knife_edge(),
sigmoid_length()
Examples
sigmoid_weight(w = c(1, 10, 100, 1000),
sigmoidal_weight = 100, sigmoidal_sigma = 3)
Derive the MizerSim marker class name for a given extension
Description
Derive the MizerSim marker class name for a given extension
Usage
simExtensionClass(extension)
Arguments
extension |
Character string — the extension (params) class name. |
Value
A character string formed by appending "Sim" to extension.
Build a MizerSim rate getter that resolves the rate functions once
Description
Internal helper capturing the pattern shared by the MizerSim rate getters
that return a species-by-size array. It validates the params and resolves the
rate functions a single time, then for each saved time step calculates only
the required target rate with mizer_rates_subset() and extracts the
element named slot from the result.
Usage
sim_size_rate(
sim,
time_range,
drop,
target,
slot,
value_name,
units = NULL,
use_sim_effort = FALSE,
representation = "point",
...
)
Arguments
sim |
A |
time_range |
Passed to the sim iteration helper. |
drop |
Passed to the sim iteration helper. |
target |
Name of the rate to calculate (as in |
slot |
Name of the element to extract from the |
value_name, units |
Metadata for the returned array. |
use_sim_effort |
If |
... |
Passed on to the rate functions. |
Value
An ArrayTimeBySpeciesBySize object (or a reduced array if drop).
Build a MizerSim rate getter that resolves the rate functions once
Description
Like sim_size_rate() but for getters that return one value per species at
each time step (a time-by-species array), such as getRDI() and getRDD().
By default these use the initial effort, matching their MizerParams counterparts.
Usage
sim_species_rate(
sim,
time_range,
target,
slot,
value_name,
units = NULL,
use_sim_effort = FALSE,
...
)
Arguments
sim |
A |
time_range |
Passed to the sim iteration helper. |
target |
Name of the rate to calculate (as in |
slot |
Name of the element to extract from the |
value_name, units |
Metadata for the returned array. |
use_sim_effort |
If |
... |
Passed on to the rate functions. |
Value
An ArrayTimeBySpecies object.
Species parameters
Description
These functions allow you to get or set the species-specific parameters stored in a MizerParams object.
Usage
species_params(object, ...)
species_params(object) <- value
is.species_params(x)
given_species_params(object, ...)
is.given_species_params(x)
given_species_params(object) <- value
calculated_species_params(params)
Arguments
object |
A MizerParams object, a MizerSim object or a data frame |
... |
Other arguments passed to methods. |
value |
A data frame with the new species parameters. |
x |
An object to test with |
params |
A MizerParams object. |
Details
There are a lot of species parameters and we will list them all below, but
most of them have sensible default values. The only required columns are
species for the species name and w_inf for its von Bertalanffy
asymptotic size. However if you have information about the values of other
parameters then you should provide them.
Three species parameters describe maximum sizes and play distinct roles:
-
w_infis the von Bertalanffy asymptotic size of an average individual. It is the required maximum-size parameter and is used to set default values forw_max,w_repro_maxandw_mat. -
w_repro_maxis the size at which a typical mature individual invests all of its available energy into reproduction, seesetReproduction(). It is not a hard ceiling on size and defaults tow_inf. -
w_maxis purely a computational boundary: it sets the upper end of the size grid and the range of plots. It defaults to1.5 * w_inf. For backwards compatibility, ifw_infis not supplied it is taken fromw_repro_maxorw_maxinstead.
Mizer distinguishes between the species parameters that you have given
explicitly and the species parameters that have been calculated by mizer or
set to default values. You can retrieve the given species parameters with
given_species_params() and the calculated ones with
calculated_species_params(). You get all species_params with
species_params().
When you change species parameters with species_params<-(), mizer
automatically detects which parameters you have changed. It records these
changed parameters in given_species_params so that they are protected
against being overwritten by future recalculations. It then triggers a
re-calculation of the calculated species parameters.
There are some species parameters that are used to set up the size-dependent parameters that are used in the mizer model:
-
gammaandqare used to set the search volume, seesetSearchVolume(). -
handnare used to set the maximum intake rate, seesetMaxIntakeRate(). -
k,ksandpare used to set activity and basic metabolic rate, seesetMetabolicRate(). -
z0,z_extanddare used to set the external mortality rate, seesetExtMort(). -
E_extandnare used to set the external encounter rate, seesetExtEncounter(). -
D_extandnare used to set the external diffusion rate, seesetExtDiffusion(). -
w_mat,w_mat25,w_repro_maxandmare used to set the allocation to reproduction, seesetReproduction(). -
pred_kernel_typespecifies the shape of the predation kernel. The default is a "lognormal", for other options see the "Setting predation kernel" section in the help forsetPredKernel(). -
betaandsigmaare parameters of the lognormal predation kernel, seelognormal_pred_kernel(). There will be other parameters if you are using other predation kernel functions.
When you change one of the above species parameters using
species_params<-() or given_species_params<-(), the new value will be
used to update the corresponding size-dependent rates automatically, unless
you have set those size-dependent rates manually, in which case the
corresponding species parameters will be ignored.
There are some species parameters that are used directly in the model rather than being used for setting up size-dependent parameters:
-
alphais the assimilation efficiency, the proportion of the consumed biomass that can be used for growth, metabolism and reproduction, see the help forgetEReproAndGrowth(). -
w_minis the egg size. -
interaction_resourcesets the interaction strength with the resource, see "Predation encounter" section in the help forgetEncounter(). -
ereprois the reproductive efficiency, the proportion of the energy invested into reproduction that is converted to egg biomass, seegetRDI(). -
R_maxis the parameter in the Beverton-Holt density dependence added to the reproduction, seesetBevertonHolt(). There will be other such parameters if you use other density dependence functions, see the "Density dependence" section in the help forsetReproduction().
Two parameters are used only by functions that need to convert between weight and length:
-
aandbare the parameters in the allometric weight-length relationshipw = a l ^ b.
If you have supplied the a and b parameters, then you can replace weight
parameters like w_inf, w_max, w_mat, w_mat25, w_repro_max and
w_min by their corresponding length parameters l_inf, l_max, l_mat,
l_mat25, l_repro_max and l_min.
The parameters that are only used to calculate default values for other parameters are:
-
f0is the feeding level and is used to get a default value for the coefficient of the search volumegamma, seeget_gamma_default(). -
fcis the critical feeding level below which the species can not maintain itself. This is used to get a default value for the coefficientksof the metabolic rate, seeget_ks_default(). -
age_matis the age at maturity and is used to get a default value for the coefficienthof the maximum intake rate, seeget_h_default(). If
age_matis not supplied, mizer used the von Bertalanffy parametersk_vb,w_infandt0as well as the weight-length exponentbto determine it. This is unreliable and is therefore not recommended.
Changing these parameters with species_params<-() will trigger a
recalculation of the downstream parameters, provided they are not protected
by being explicitly given.
There are other species parameters that are used in tuning the model to observations:
-
biomass_observedandbiomass_cutoffallow you to specify for each species the total observed biomass above some cutoff size. This is used bycalibrateBiomass()andmatchBiomasses(). -
yield_observedallows you to specify for each species the total annual fisheries yield. This is used bycalibrateYield()andmatchYields().
Finally there are two species parameters that control the way the species are represented in plots:
-
linecolourspecifies the colour and can be any valid R colour value. -
linetypespecifies the line type ("solid", "dashed", "dotted", "dotdash", "longdash", "twodash" or "blank")
Other species-specific information that is related to how the species is
fished is specified in a gear parameter data frame, see gear_params().
However in the case where each species is caught by only a single gear,
this information can also optionally be provided as species parameters and
newMultispeciesParams() will transfer them to the gear_params data frame.
However changing these parameters later in the species parameter data frames
will have no effect.
You are allowed to include additional columns in the species parameter data frames. They will simply be ignored by mizer but will be stored in the MizerParams object, in case your own code makes use of them.
Value
species_params(): Data frame containing all species parameters
currently stored in the model.
species_params<-(): Updates the given_species_params with any
parameters you have changed, and then recalculates the full species
parameter table and the model parameters.
given_species_params(): Data frame containing the species parameter
values that were supplied explicitly by the user.
given_species_params<-(): An alternative to species_params<-() that
also triggers a recalculation of other parameters. The only difference is
that given_species_params<-() issues warnings when a parameter is
changed whose effect is overridden by another parameter that has already
been given. This is especially useful during interactive use.
calculated_species_params(): Data frame containing only those species
parameter entries that are not explicit user input. Columns that would
consist entirely of NA values are dropped.
is.species_params() returns TRUE if x is a species_params
object, FALSE otherwise.
is.given_species_params() returns TRUE if x is a
given_species_params object, FALSE otherwise.
See Also
validSpeciesParams(), setParams()
Other functions for setting parameters:
gear_params(),
setExtDiffusion(),
setExtEncounter(),
setExtMort(),
setFishing(),
setInteraction(),
setMaxIntakeRate(),
setMetabolicRate(),
setParams(),
setPredKernel(),
setReproduction(),
setSearchVolume(),
use_predation_diffusion()
Look up the size-bin widths for spectra data
Description
Matches the weights in the plotting data to the model's full size grid and returns the corresponding bin widths.
Usage
spectra_bin_width(w, params)
Arguments
w |
Numeric vector of weights. |
params |
A MizerParams object. |
Value
A numeric vector of bin widths, one for each entry of w.
Y-axis label for a size-spectrum plot
Description
Y-axis label for a size-spectrum plot
Usage
spectra_y_label(power)
Arguments
power |
The power of weight that the abundance was multiplied by. |
Value
A character string for the y-axis label.
Set initial values to a steady state for the model
Description
The steady state is found by running the dynamics while keeping reproduction,
resource and other components constant until the size spectra no longer
change much (or until time t_max is reached, if earlier).
Usage
steady(
params,
t_max = 100,
t_per = 1.5,
dt = 0.1,
tol = 0.1 * dt,
return_sim = FALSE,
preserve = c("reproduction_level", "erepro", "R_max"),
progress_bar = TRUE,
info_level = 3,
method = c("euler", "predictor_corrector", "tr_bdf2")
)
Arguments
params |
A MizerParams object |
t_max |
The maximum number of years to run the simulation. Default is 100. |
t_per |
The simulation is broken up into shorter runs of |
dt |
The time step to use in |
tol |
The simulation stops when the relative change in the egg
production RDI over |
return_sim |
If TRUE, the function returns the MizerSim object holding
the result of the simulation run, saved at intervals of |
preserve |
|
progress_bar |
A shiny progress object to implement a progress bar in a shiny app. Default FALSE. |
info_level |
Controls the amount of information messages that are shown. Higher levels lead to more messages. |
method |
The numerical method to use for the consumer density update.
See |
Details
If the model use Beverton-Holt reproduction then the reproduction parameters
are set to values that give the level of reproduction observed in that
steady state. The preserve argument can be used to specify which of the
reproduction parameters should be preserved.
The resource is rebalanced so that the returned state is a steady state of the resource as well as of the consumers. The resource abundance is preserved while the capacity is recomputed to keep it steady. If the resource capacity had been set manually (frozen), it is rebalanced and thereby unfrozen.
Value
If return_sim = FALSE, a MizerParams object with the initial
state replaced by the steady state. If return_sim = TRUE, a MizerSim
object containing the intermediate states saved every t_per years.
Examples
params <- newTraitParams()
species_params(params)$gamma[5] <- 3000
params <- steady(params)
plotSpectra(params)
Set initial abundances to solution of steady-state equation with current rates
Description
This first calculates growth and death rates that arise from the current
initial abundances. Then it solves the steady-state equation with these
growth and death rates and the current abundance at the smallest size.
It sets the initial abundances of the selected species to this solution.
Usage
steadySingleSpecies(
params,
species = NULL,
keep = c("egg", "biomass", "number")
)
Arguments
params |
A MizerParams object |
species |
The species to be selected. Optional. By default all target species are selected. A vector of species names, or a numeric vector with the species indices, or a logical vector indicating for each species whether it is to be selected (TRUE) or not. |
keep |
A string determining which quantity is to be kept constant. The choices are "egg" which keeps the egg density constant, "biomass" which keeps the total biomass of the species constant and "number" which keeps the total number of individuals constant. |
Details
The function only changes the initial abundances. It does not adjust the reproduction parameters or any other parameters. Therefore the result of applying this function is of course not a steady state, because after changing the abundances of the selected species the growth, death and reproduction rates will have changed.
If the keep argument is supplied, the solution for the selected species
are rescaled to keep the specified quantity at the value they had before
calling this function.
Value
A MizerParams object in which the initial abundances of the selected species are changed to their single-species steady state abundances.
Examples
# Set initial abundance of Cod to its single-species steady state
params <- steadySingleSpecies(NS_params, species = "Cod")
Display the structure of mizer objects
Description
Mizer provides str() methods for MizerParams() and MizerSim() objects,
as well as ArraySpeciesBySize(), ArrayTimeBySpecies() and ArrayTimeBySpeciesBySize()
objects. These methods produce a clean, compact overview of the object's structure
without polluting the console with large amounts of internal data.
Usage
## S3 method for class 'ArraySpeciesBySize'
str(object, ...)
## S3 method for class 'ArrayTimeBySpecies'
str(object, ...)
## S3 method for class 'ArrayTimeBySpeciesBySize'
str(object, ...)
## S3 method for class 'MizerSim'
str(object, max.level = NA, ...)
## S3 method for class 'MizerParams'
str(object, max.level = NA, ...)
Arguments
object |
The object to display the structure of. |
max.level |
Maximum level of nesting to print. Defaults to |
... |
Further arguments. They are passed to the default |
Value
NULL, invisibly.
See Also
print(), as.data.frame(), summary(), plot(), MizerParams(), MizerSim(), ArraySpeciesBySize(),
ArrayTimeBySpecies(), ArrayTimeBySpeciesBySize()
Examples
str(NS_params)
str(NS_sim)
str(getEncounter(NS_params))
Summarise mizer objects
Description
Mizer provides summary() methods for model objects and for the specialised
array classes returned by many mizer functions.
Usage
## S3 method for class 'ArraySpeciesBySize'
summary(object, ...)
## S3 method for class 'ArrayTimeBySpecies'
summary(object, ...)
## S3 method for class 'ArrayTimeBySpeciesBySize'
summary(object, ...)
## S3 method for class 'MizerSim'
summary(object, ...)
## S3 method for class 'MizerParams'
summary(object, ...)
Arguments
object |
The object to summarise. |
... |
Further arguments. They are currently ignored by the mizer methods. |
Details
For a MizerParams() object, summary() prints the model metadata, size
grids, selected species parameters and fishing gear details. For a
MizerSim() object, it first prints the parameter summary and then reports
the simulated time period and output interval.
For ArraySpeciesBySize(), ArrayTimeBySpecies() and
ArrayTimeBySpeciesBySize() objects, summary() returns a small list with
the value name, units, dimensions and a per-species data frame containing
minimum, mean and maximum values. Printing that summary object gives the same
compact table in a human-readable form.
Value
For MizerParams() and MizerSim(), the object is returned invisibly.
For array objects, a list of class summary.ArraySpeciesBySize,
summary.ArrayTimeBySpecies or summary.ArrayTimeBySpeciesBySize.
See Also
print(), as.data.frame(), str(), MizerParams(), MizerSim(),
ArraySpeciesBySize(), ArrayTimeBySpecies(),
ArrayTimeBySpeciesBySize()
Examples
summary(NS_params)
summary(NS_sim)
summary(getEncounter(NS_params))
summary(getFMort(NS_sim))
Description of summary functions
Description
Mizer provides a range of functions to summarise the results of a simulation.
Details
A list of available summary functions is given in the table below.
| Function | Returns | Description |
getDiet() | Three dimensional array (predator x size x prey) | Diet of predator at size, resolved by prey species |
getTrophicLevel() | ArraySpeciesBySize (species x size) | Trophic level of individuals at size, accounting for ontogenetic diet shifts |
getTrophicLevelBySpecies() | Named vector (species) | Consumption-rate-weighted mean trophic level of each species |
getSSB() | Two dimensional array (time x species) | Total Spawning Stock Biomass (SSB) of each species through time where SSB is calculated as the sum of weight of all mature individuals. |
getBiomass() | Two dimensional array (time x species) | Total biomass of each species through time. |
getN() | Two dimensional array (time x species) | Total abundance of each species through time. |
getFeedingLevel() | Three dimensional array (time x species x size) | Feeding level of each species by size through time. |
getM2 | Three dimensional array (time x species x size) | The predation mortality imposed on each species by size through time. |
getFMort() | Three dimensional array (time x species x size) | Total fishing mortality on each species by size through time. |
getFMortGear() | Four dimensional array (time x gear x species x size) | Fishing mortality on each species by each gear at size through time. |
getYieldGear() | Three dimensional array (time x gear x species) | Total yield by gear and species through time. |
getYield() | Two dimensional array (time x species) | Total yield of each species across all gears through time. |
See Also
indicator_functions, plotting_functions
Truncated lognormal predation kernel
Description
This is like the lognormal_pred_kernel() but with an imposed maximum
predator/prey mass ratio
Usage
truncated_lognormal_pred_kernel(ppmr, beta, sigma)
Arguments
ppmr |
A vector of predator/prey size ratios |
beta |
The preferred predator/prey size ratio |
sigma |
The width parameter of the log-normal kernel |
Details
Writing the predator mass as w and the prey mass as w_p,
the feeding kernel is given as
\phi_i(w, w_p) =
\exp \left[ \frac{-(\ln(w / w_p / \beta_i))^2}{2\sigma_i^2} \right]
if w/w_p is between 1 and
\beta_i\exp(3\sigma_i)
and zero otherwise. Here \beta_i is the preferred predator-prey mass
ratio and \sigma_i determines the width of the kernel. These two
parameters need to be given in the species parameter dataframe in the columns
beta and sigma.
This function is called from setPredKernel() to set up the
predation kernel slots in a MizerParams object.
Value
A vector giving the value of the predation kernel at each of the
predator/prey mass ratios in the ppmr argument.
See Also
Other predation kernel:
box_pred_kernel(),
lognormal_pred_kernel(),
power_law_pred_kernel()
Examples
params <- NS_params
species_params(params)$pred_kernel_type <- "truncated_lognormal"
plot(w_full(params), getPredKernel(params)["Cod", 10, ], type="l", log="x")
Upgrade the core slots of a MizerParams object
Description
This is the MizerParams method of the upgrade() generic and performs the
core mizer upgrade only. It is called from the orchestrator
runExtensionUpgrades(), which also invokes any registered extension upgrade
methods.
You should never need to call it directly; use validParams() (or
readParams()) instead.
Usage
## S3 method for class 'MizerParams'
upgrade(object, ...)
Arguments
object |
An old MizerParams object to be upgraded |
... |
Unused. |
Value
The upgraded MizerParams object
See Also
Upgrade a MizerSim object from earlier versions
Description
This is the MizerSim method of the upgrade() generic. It rebuilds the
simulation around an upgraded params object; the params upgrade (core mizer
and any extensions) is performed by the validParams() call below. You
should never need to call it directly; use validSim() (or readSim()).
Usage
## S3 method for class 'MizerSim'
upgrade(object, ...)
Arguments
object |
An old MizerSim object to be upgraded |
... |
Unused. |
Value
The upgraded MizerSim object
Back-compatible wrapper for the core MizerParams upgrade
Description
Retained because it was an (unexported) internal entry point. New code should
rely on validParams() / readParams(), which orchestrate the core upgrade
together with any extension upgrades.
Usage
upgradeParams(params)
Arguments
params |
An old MizerParams object. |
Value
The upgraded MizerParams object.
Back-compatible wrapper for the MizerSim upgrade
Description
Retained because it was an (unexported) internal entry point. New code should
rely on validSim() / readSim().
Usage
upgradeSim(sim)
Arguments
sim |
An old MizerSim object. |
Value
The upgraded MizerSim object.
Get or set the use_predation_diffusion flag
Description
Controls whether predation-induced diffusion is included when calculating
rates with mizerDiffusion(). When FALSE (the default), the
predation-driven diffusion term is omitted, preserving the behaviour of
previous mizer versions. Set to TRUE to enable the diffusion term from
the jump-growth equation.
Usage
use_predation_diffusion(params)
use_predation_diffusion(params) <- value
Arguments
params |
A MizerParams object. |
value |
A single logical value ( |
Value
use_predation_diffusion(): A single logical value.
use_predation_diffusion<-: A MizerParams object with the
use_predation_diffusion flag updated.
See Also
Other functions for setting parameters:
gear_params(),
setExtDiffusion(),
setExtEncounter(),
setExtMort(),
setFishing(),
setInteraction(),
setMaxIntakeRate(),
setMetabolicRate(),
setParams(),
setPredKernel(),
setReproduction(),
setSearchVolume(),
species_params()
Test whether a mizer object uses extension S4 dispatch
Description
Test whether a mizer object uses extension S4 dispatch
Usage
usesExtensionDispatch(object)
Arguments
object |
A |
Value
TRUE if the object's primary class is not the plain base class.
Make a valid effort vector
Description
Make a valid effort vector
Usage
validEffortVector(effort, params)
Arguments
effort |
A vector or scalar with the initial fishing effort, see Details below. |
params |
A MizerParams object. |
Details
A valid effort vector is a named vector with one effort value for each gear. However you can also supply the effort value in different ways:
a scalar, which is then replicated for each gear
an unnamed vector, which is then assumed to be in the same order as the gears in the params object
a named vector in which the gear names have a different order than in the params object. This is then sorted correctly.
a named vector which only supplies values for some of the gears. The effort for the other gears is then set to the default effort returned by
validEffortVector(), which depends on the defaults edition.
These conversions are done by the function validEffortVector().
An effort argument will lead to an error if it is either
unnamed and of the wrong length
named but where some names do not match any of the gears
not numeric
See Also
Check validity of gear parameters and set defaults
Description
The function returns a valid gear parameter data frame that can be used
by setFishing() or it gives an error message.
Usage
validGearParams(gear_params, species_params)
Arguments
gear_params |
Gear parameter data frame |
species_params |
Species parameter data frame |
Details
The gear_params data frame is allowed to have zero rows, but if it has rows, then the following requirements apply:
There must be columns
speciesandgearand any species - gear pair is allowed to appear at most once. Any species that appears must also appear in thespecies_paramsdata frame.There must be a
sel_funccolumn. If a selectivity function is not supplied, it will be set to "knife_edge".There must be a
catchabilitycolumn. If a catchability is not supplied, it will be set to 1.All the parameters required by the selectivity functions must be provided.
If gear_params is empty, then this function tries to find the necessary information in the species_params data frame. This restricts each species to be fished by only one gear. Defaults are used for information that can not be found in the species_params dataframe, as follows:
If there is no
gearcolumn or it is NA then a new gear named after the species is introduced.If there is no
sel_funccolumn or it is NA thenknife_edgeis used.If there is no
catchabilitycolumn or it is NA then this is set to 1.If the selectivity function is
knife_edgeand noknife_edge_sizeis provided, it is set tow_mat.
The row names of the returned data frame are of the form "species, gear".
When gear_params is NULL and there is no gear information in
species_params, then a gear called knife_edge_gear is set up with a
knife_edge selectivity for each species and a knive_edge_size equal to
w_mat. Catchability is set to 0.3 for all species.
Value
A valid gear parameter data frame
See Also
Validate MizerParams object and upgrade if necessary
Description
Checks that the given MizerParams object is valid and upgrades it if necessary.
Usage
validParams(params, info_level = 3)
Arguments
params |
The MizerParams object to validate |
info_level |
Controls the amount of information messages and warnings that are shown. Higher levels lead to more messages. |
Details
It is possible to render a MizerParams object invalid by manually changing its slots. This function checks that the object is valid and if not it attempts to upgrade it to a valid object or gives an error message. If the object is valid then it is returned unchanged. The function reports an error if any of the rate arrays contain any non-finite numbers (except for the maximum intake rate that is allowed to be infinite).
Occasionally, during the development of new features for mizer, the
MizerParams object gains extra slots. MizerParams objects
created in older versions of mizer are then no longer valid in the new
version because of the missing slots. You need to upgrade them with this
function. It adds the missing slots and fills them with default values. Any
object from version 0.4 onwards can be upgraded. Any old
MizerSim objects should be similarly updated with
validSim().
This function uses newMultispeciesParams() to create a new
MizerParams object using the parameters extracted from the old MizerParams
object.
Value
A valid MizerParams object
Backwards compatibility
The internal numerics in mizer have changed over time, so there may be small discrepancies between the results obtained with the upgraded object in the new version and the original object in the old version. If it is important for you to reproduce the exact results then you should install the version of mizer with which you obtained the results. You can do this with
remotes::install_github("sizespectrum/mizer", ref = "v0.2")
where you should replace "v0.2" with the version number you require. You can see the list of available releases at https://github.com/sizespectrum/mizer/tags.
If you only have a serialised version of the old object, for example
created via saveRDS(), and you get an error when trying to read it in
with readRDS() then unfortunately you will need to install the old version
of mizer first to read the params object into your workspace, then switch
to the current version and then call validParams(). You can then save
the new version again with saveParams().
Validate MizerSim object and upgrade if necessary
Description
Checks that the given MizerSim object is valid and upgrades it if necessary.
It also validates the embedded MizerParams-class() object with
validParams(). If any entries of the consumer abundance array sim@n are
non-finite, a warning is issued and the simulation is truncated at the last
time step where sim@n is still finite.
Usage
validSim(sim)
Arguments
sim |
The MizerSim object to validate |
Details
Occasionally, during the development of new features for mizer, the MizerSim class or the MizerParams class gains extra slots. MizerSim objects created in older versions of mizer are then no longer valid in the new version because of the missing slots. You need to upgrade them with this function.
This function adds the missing slots and fills them with default values. It
also calls validParams() to upgrade the MizerParams object inside the
MizerSim object. Any object from version 0.4 onwards can be upgraded.
Value
A valid MizerSim object
Backwards compatibility
The internal numerics in mizer have changed over time, so there may be small discrepancies between the results obtained with the upgraded object in the new version and the original object in the old version. If it is important for you to reproduce the exact results then you should install the version of mizer with which you obtained the results. You can do this with
remotes::install_github("sizespectrum/mizer", ref = "v0.2")
where you should replace "v0.2" with the version number you require. You can see the list of available releases at https://github.com/sizespectrum/mizer/tags.
If you only have a serialised version of the old object, for example
created via saveRDS(), and you get an error when trying to read it in
with readRDS() then unfortunately you will need to install the old version
of mizer first to read the params object into your workspace, then switch
to the current version and then call validParams(). You can then save
the new version again with saveParams().
Validate species parameter data frame
Description
These functions check the validity of a species parameter frame and, where
necessary, make corrections. validGivenSpeciesParams() only checks and
corrects the given species parameters but does not add default values for
species parameters that were not provided. validSpeciesParams() first calls
validGivenSpeciesParams() but then goes further by adding default values
for species parameters that were not provided.
Usage
validSpeciesParams(species_params)
validGivenSpeciesParams(species_params)
Arguments
species_params |
The user-supplied species parameter data frame |
Details
validGivenSpeciesParams() checks the validity of the given species
parameters. It throws an error if
the
speciescolumn does not exist or contains duplicatesthe asymptotic size
w_infis not specified for all species (but see the backwards-compatibility note below)
If a weight-based parameter is missing but the corresponding length-based
parameter is given, as well as the a and b parameters for length-weight
conversion, then the weight-based parameters are added. If both length and
weight are given, then weight is used and an info_about_default condition
is signalled if the two are inconsistent.
The required maximum-size parameter is w_inf, the von Bertalanffy
asymptotic size of an average individual. For backwards compatibility, if no
w_inf column is given, its values are taken from the w_repro_max column
if that is present, or otherwise from the w_max column, and an
informational message is issued. (w_repro_max is preferred over w_max
because in earlier versions of mizer it was the size at which growth stopped
and is therefore the closest analogue to the asymptotic size.)
Some inconsistencies in the size parameters are resolved as follows:
Any
w_matthat is not smaller thanw_infis set tow_inf / 4.Any
w_mat25that is not smaller thanw_matis set to NA.Any
w_minthat is not smaller thanw_matis set to0.001orw_mat /10, whichever is smaller.Any
w_repro_maxthat is not larger thanw_matis set to4 * w_mat.
The row names of the returned data frame will be the species names.
If species_params was provided as a tibble it is converted back to an
ordinary data frame.
The function tests for some typical misspellings of parameter names, like wrong capitalisation or missing underscores and issues a warning if it detects such a name.
validSpeciesParams() first calls validGivenSpeciesParams() but then
goes further by adding default values for species parameters that were not
provided. It only sets defaults for those species parameters that are not
owned by a single rate-setting function, namely those that are read by
several of them (n), that are used only when projecting (alpha), that
determine the size grid (w_min, w_max), that are needed for the
length-weight conversion (a, b) or that are used only for reporting
(is_background). The function sets default values if any of the following
species parameters are missing or NA:
-
w_maxis set to1.5 * w_inf(it is only a computational boundary) -
w_repro_maxis set tow_inf -
w_matis set tow_inf/4 -
w_minis set to0.001 -
alphais set to0.6 -
nis set to3/4 -
ais set to0.01 -
bis set to3 -
is_backgroundis set toFALSE
All other species parameters are given their default values by the
rate-setting function that uses them, so that each default has a single
home. For example p and k are set by setMetabolicRate(), z_ext, d
and z0 by setExtMort(), E_ext by setExtEncounter(), D_ext by
setExtDiffusion(), interaction_resource by setInteraction(), beta
and sigma by setPredKernel(), q and gamma by setSearchVolume(),
and erepro, m, w_mat25 and R_max by setReproduction(). These
columns are therefore absent from the data frame returned by
validSpeciesParams() but present in the species parameters of a
MizerParams object, because setParams() calls all the rate-setting
functions.
Note that the species parameters returned by these functions are not
guaranteed to produce a viable model. More checks of the parameters are
performed by the individual rate-setting functions (see setParams() for the
list of these functions).
Value
For validSpeciesParams(): A valid species parameter data frame with
additional parameters with default values.
For validGivenSpeciesParams(): A valid species parameter data frame
without additional parameters.
See Also
species_params(), validGearParams(), validParams(), validSim()
Helper function to assure validity of gears argument
Description
If the gears argument contains invalid gears, then these are ignored but a warning is issued.
Usage
valid_gears_arg(object, gears = NULL, error_on_empty = FALSE)
Arguments
object |
A MizerSim or MizerParams object from which the gears should be selected. |
gears |
The gears to be selected. Optional. By default all gears are selected. A vector of gear names. |
error_on_empty |
Whether to throw an error if there are zero valid gears. Default FALSE. |
Value
A vector of gear names in the same order as supplied in gears,
with invalid names removed. If gears is NULL, all gears are returned
in the order stored in the model.
Helper function to assure validity of species argument
Description
If the species argument contains invalid species, then these are ignored but a warning is issued.
Usage
valid_species_arg(
object,
species = NULL,
return.logical = FALSE,
error_on_empty = FALSE
)
Arguments
object |
A MizerSim or MizerParams object from which the species should be selected. |
species |
The species to be selected. Optional. By default all target species are selected. A vector of species names, or a numeric vector with the species indices, or a logical vector indicating for each species whether it is to be selected (TRUE) or not. |
return.logical |
Whether the return value should be a logical vector. Default FALSE. |
error_on_empty |
Whether to throw an error if there are zero valid species. Default FALSE. |
Value
A vector of species names, in the same order as specified in the 'species' argument. If 'return.logical = TRUE' then a logical vector is returned instead, with length equal to the number of species, with TRUE entry for each selected species.
Validate and normalise an extensions named character vector
Description
Checks that extensions is a named character vector with unique,
syntactically valid names, and normalises NULL to character().
Usage
validateExtensionsVector(extensions)
Arguments
extensions |
A named character vector, or |
Value
A validated named character vector (possibly length-zero).
Apply a second_order_w value to the current slot list
Description
Internal helper that validates a second_order_w value (a single logical,
a single flux scheme name, or a named vector with entries flux and/or
bin_average) and returns the updated named list. Shared by the
second_order_w<- setter and by the model constructors (e.g.
newMultispeciesParams()), which set the slot directly before the rest of
the parameters are computed so that the bin-averaged constructions pick up
the flag, without the setter's extra setParams() call.
Usage
validate_second_order_w(current, value)
Arguments
current |
The current |
value |
The value to apply, as described above. |
Value
The updated second_order_w list.
Size bins
Description
Functions to fetch information about the size bins used in the model
described by params.
Usage
w(params)
w_full(params)
dw(params)
dw_full(params)
Arguments
params |
A MizerParams object |
Details
To represent the continuous size spectrum in the computer, the size
variable is discretized into a vector w of discrete weights,
providing a grid of sizes spanning the range from the smallest egg size
to the largest maximum size. These grid values divide the full size
range into a finite number of size bins. The size bins should be chosen
small enough to avoid the discretisation errors from becoming too big.
You can fetch this vector with w() and the vector of bin widths with
dw().
The weight grid is set up to be logarithmically spaced, so that
w[j]=w[1]*10^(j*dx) for some fixed dx. This means that the bin widths
increase with size: dw[j] = w[j] * (10^dx - 1).
This grid is set up automatically when creating a MizerParams object.
Because the resource spectrum spans a larger range of sizes, these sizes
are discretized into a different vector of weights w_full. This usually
starts at a much smaller size than w, but also runs up to the same
largest size, so that the last entries of w_full have to coincide with the
entries of w. The logarithmic spacing for w_full is the same as that for
w, so that again w_full[j]=w_full[1]*10^(j*dx). The function w_full()
gives the vector of sizes and dw_full() gives the vector of bin widths.
You will need these vectors when converting number densities to numbers.
For example the size spectrum of a species is stored as a vector of
values that represent the density of fish in each size bin rather than
the number of fish. The number of fish in the size bin between w[j] and
w[j+1]=w[j]+dw[j] is obtained as N[j]*dw[j].
The vector w can be used for example to convert the number of individuals
in a size bin into the biomass in the size bin. The biomass in the
jth bin is biomass[j] = N[j] * dw[j] * w[j].
Of course all these calculations with discrete sizes and size bins are
only giving approximations to the continuous values, and these approximations
get better the smaller the size bins are, i.e., the more size bins are
used. However using more size bins also slows down the calculations, so
there is a trade-off. This is why the functions setting up MizerParams
objects allow you to choose the number of size bins no_w.
Value
w() returns a vector with the sizes at the start of each size bin
of the consumer spectrum.
w_full() returns a vector with the sizes at the start of each size
bin of the resource spectrum, which typically starts at smaller sizes than
the consumer spectrum.
dw() returns a vector with the widths of the size bins of the
consumer spectrum.
dw_full() returns a vector with the widths of the size bins of the
resource spectrum.
Examples
str(w(NS_params))
str(dw(NS_params))
str(w_full(NS_params))
str(dw_full(NS_params))
# Calculating the biomass of Cod in each bin in the North Sea model
biomass <- initialN(NS_params)["Cod", ] * dw(NS_params) * w(NS_params)
# Summing to get total biomass
sum(biomass)